Feyzi Sinan Tokalı, Halil Şenol, Şeyma Ateşoğlu, Fahri Akbaş
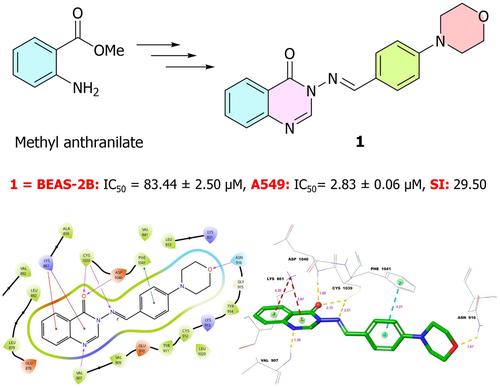
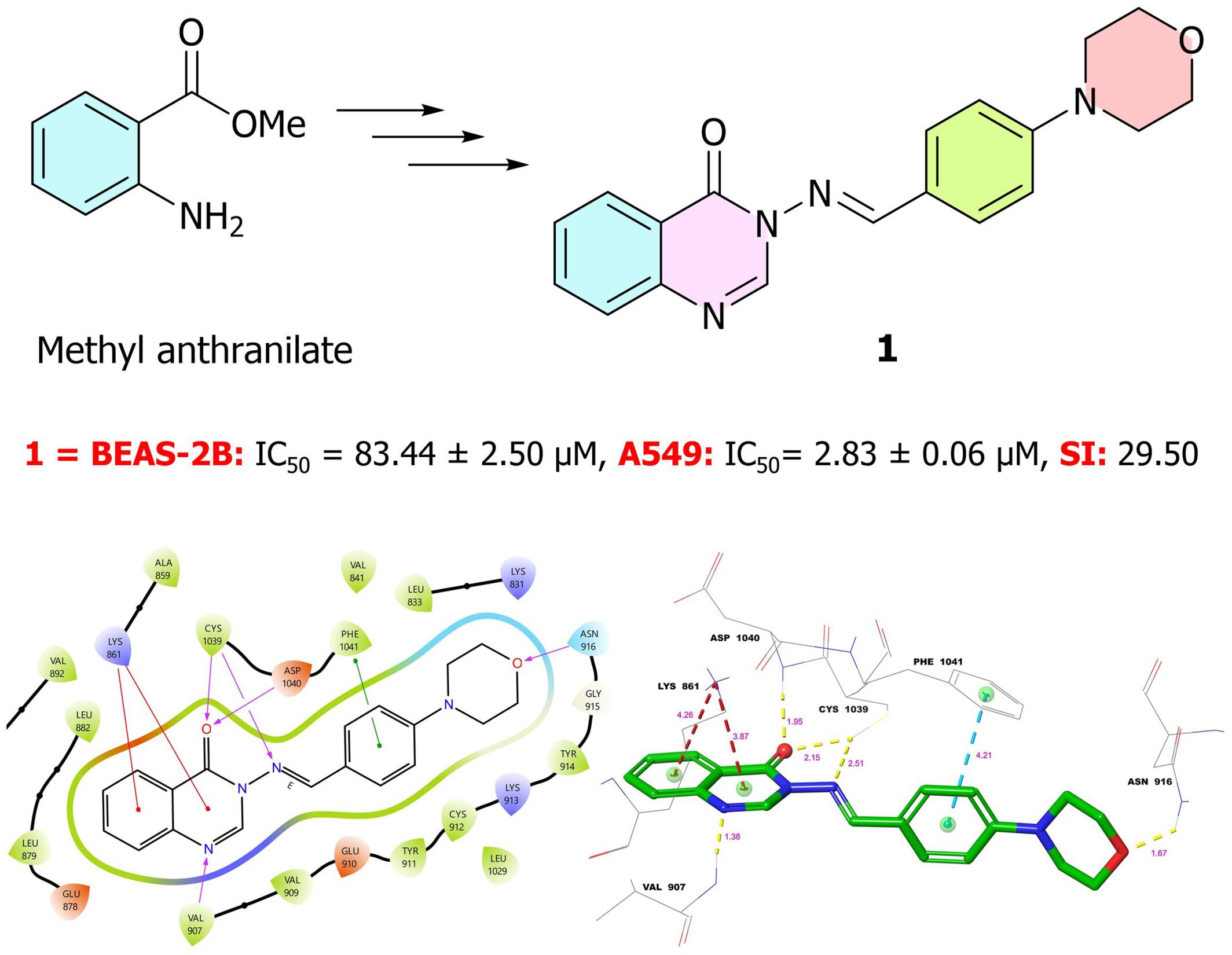
{"title":"作为抗肺癌药物的一系列喹唑啉-4(3H)-酮-吗啉混合物:合成、分子对接、分子动力学、ADME 预测和生物活性研究。","authors":"Feyzi Sinan Tokalı, Halil Şenol, Şeyma Ateşoğlu, Fahri Akbaş","doi":"10.1111/cbdd.14599","DOIUrl":null,"url":null,"abstract":"<p>In this study, we synthesized 15 novel quinazoline-morpholinobenzylideneamino hybrid compounds from methyl anthranilate and we assessed their cytotoxicity via in vitro assays against A549 and BEAS-2B cell lines. Molecular docking studies were conducted to evaluate the protein-ligand interactions and inhibition mechanisms on nine different molecular targets, while molecular dynamics (MD) simulations were carried out to assess the stability of the best docked ligand–protein complexes. Additionally, ADME prediction was carried out to determine physicochemical parameters and drug likeness. According to the cytotoxicity assays, compound <b>1</b> (IC<sub>50</sub> = 2.83 μM) was found to be the most active inhibitor against A549 cells. While the selectivity index (SI) of compound <b>1</b> is 29, the SI of the reference drugs paclitaxel and sorafenib, used in this study, are 2.40 and 4.92, respectively. Among the hybrid compounds, <b>1</b> has the best docking scores against VEGFR1 (−11.744 kcal/mol), VEGFR2 (−12.407 kcal/mol) and EGFR (−10.359 kcal/mol). During MD simulations, compound <b>1</b> consistently exhibited strong hydrogen bond interactions with the active sites of VEGFR1 and 2, and these interactions were maintained for more than 90% of the simulation time. Additionally, the RMSD and RMSF values of the ligand–protein complexes exhibited high stability at their minimum levels around 1–2 Å. In conclusion, these findings suggest that compound <b>1</b> may be a potent and selective inhibitor candidate for lung cancer treatment and inhibition of VEGFR2, especially.</p>","PeriodicalId":143,"journal":{"name":"Chemical Biology & Drug Design","volume":"104 1","pages":""},"PeriodicalIF":3.2000,"publicationDate":"2024-07-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"A series of quinazolin-4(3H)-one-morpholine hybrids as anti-lung-cancer agents: Synthesis, molecular docking, molecular dynamics, ADME prediction and biological activity studies\",\"authors\":\"Feyzi Sinan Tokalı, Halil Şenol, Şeyma Ateşoğlu, Fahri Akbaş\",\"doi\":\"10.1111/cbdd.14599\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>In this study, we synthesized 15 novel quinazoline-morpholinobenzylideneamino hybrid compounds from methyl anthranilate and we assessed their cytotoxicity via in vitro assays against A549 and BEAS-2B cell lines. Molecular docking studies were conducted to evaluate the protein-ligand interactions and inhibition mechanisms on nine different molecular targets, while molecular dynamics (MD) simulations were carried out to assess the stability of the best docked ligand–protein complexes. Additionally, ADME prediction was carried out to determine physicochemical parameters and drug likeness. According to the cytotoxicity assays, compound <b>1</b> (IC<sub>50</sub> = 2.83 μM) was found to be the most active inhibitor against A549 cells. While the selectivity index (SI) of compound <b>1</b> is 29, the SI of the reference drugs paclitaxel and sorafenib, used in this study, are 2.40 and 4.92, respectively. Among the hybrid compounds, <b>1</b> has the best docking scores against VEGFR1 (−11.744 kcal/mol), VEGFR2 (−12.407 kcal/mol) and EGFR (−10.359 kcal/mol). During MD simulations, compound <b>1</b> consistently exhibited strong hydrogen bond interactions with the active sites of VEGFR1 and 2, and these interactions were maintained for more than 90% of the simulation time. Additionally, the RMSD and RMSF values of the ligand–protein complexes exhibited high stability at their minimum levels around 1–2 Å. In conclusion, these findings suggest that compound <b>1</b> may be a potent and selective inhibitor candidate for lung cancer treatment and inhibition of VEGFR2, especially.</p>\",\"PeriodicalId\":143,\"journal\":{\"name\":\"Chemical Biology & Drug Design\",\"volume\":\"104 1\",\"pages\":\"\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2024-07-22\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Chemical Biology & Drug Design\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/cbdd.14599\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Chemical Biology & Drug Design","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/cbdd.14599","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
A series of quinazolin-4(3H)-one-morpholine hybrids as anti-lung-cancer agents: Synthesis, molecular docking, molecular dynamics, ADME prediction and biological activity studies
In this study, we synthesized 15 novel quinazoline-morpholinobenzylideneamino hybrid compounds from methyl anthranilate and we assessed their cytotoxicity via in vitro assays against A549 and BEAS-2B cell lines. Molecular docking studies were conducted to evaluate the protein-ligand interactions and inhibition mechanisms on nine different molecular targets, while molecular dynamics (MD) simulations were carried out to assess the stability of the best docked ligand–protein complexes. Additionally, ADME prediction was carried out to determine physicochemical parameters and drug likeness. According to the cytotoxicity assays, compound 1 (IC50 = 2.83 μM) was found to be the most active inhibitor against A549 cells. While the selectivity index (SI) of compound 1 is 29, the SI of the reference drugs paclitaxel and sorafenib, used in this study, are 2.40 and 4.92, respectively. Among the hybrid compounds, 1 has the best docking scores against VEGFR1 (−11.744 kcal/mol), VEGFR2 (−12.407 kcal/mol) and EGFR (−10.359 kcal/mol). During MD simulations, compound 1 consistently exhibited strong hydrogen bond interactions with the active sites of VEGFR1 and 2, and these interactions were maintained for more than 90% of the simulation time. Additionally, the RMSD and RMSF values of the ligand–protein complexes exhibited high stability at their minimum levels around 1–2 Å. In conclusion, these findings suggest that compound 1 may be a potent and selective inhibitor candidate for lung cancer treatment and inhibition of VEGFR2, especially.
期刊介绍:
Chemical Biology & Drug Design is a peer-reviewed scientific journal that is dedicated to the advancement of innovative science, technology and medicine with a focus on the multidisciplinary fields of chemical biology and drug design. It is the aim of Chemical Biology & Drug Design to capture significant research and drug discovery that highlights new concepts, insight and new findings within the scope of chemical biology and drug design.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


