Daoyang Zhang, Lauren E. Rosch, Matthew R. Crawley and Timothy R. Cook
{"title":"对双铁(III)-μ-氧卟啉棱柱进行后合成修饰以增强氧还原电催化性能","authors":"Daoyang Zhang, Lauren E. Rosch, Matthew R. Crawley and Timothy R. Cook","doi":"10.1039/D4QI01219D","DOIUrl":null,"url":null,"abstract":"<p >Bis-iron(<small>III</small>)-μ-oxo porphyrins are known electrocatalysts for Oxygen Reduction Reaction (ORR) that favor four-electron four-proton chemistry. The facile formation of the μ-oxo motif makes forming cofacial structures straightforward unlike for other metalloporphyrins where the face-to-face geometry must be enforced by tethering groups. We have pioneered the use of coordination chemistry to obtain cofacial porphyrin ORR catalysts containing Co(<small>II</small>) ions. Here, we adapt our use of molecular clips for the post-synthetic modification of μ-oxo porphyrin dimers rather than as self-assembly building blocks. Although the cofacial geometry is inherent to the μ-oxo core, under ORR catalysis conditions, the dimer is rapidly cleaved, resulting in reactivity differences between traditional unclipped bis-iron(<small>III</small>)-μ-oxo porphyrins and our post-synthetically tethered architectures. We demonstrate our approach using two molecular clips that differ in length. The bis-iron(<small>III</small>)-μ-oxo precatalysts were characterized by <small><sup>1</sup></small>H NMR, ESI-MS, and molecular modeling to support the formation of cofacial prisms. Catalytic activity was studied electrochemically using cyclic voltammetry and hydrodynamic voltammetry. Whereas untethered bis-iron(<small>III</small>)-μ-oxo porphyrins are limited to heterogenous catalysis so that the dimeric structure is not lost upon the removal of the oxo bridge, our clipped architectures show significant catalytic current response under homogeneous conditions. Furthermore, when a shorter molecular clip is used as a post-synthetic tether, the selectivity under heterogeneous conditions is significantly enhanced: monomeric Fe(<small>III</small>) tetraphenylporphyrin (TPhP) generates 64.3% H<small><sub>2</sub></small>O<small><sub>2</sub></small>. When this same porphyrin is templated into a cofacial environment (Fe<small><sub>2</sub></small>O TPhP) the selectivity improves to 15.8% H<small><sub>2</sub></small>O<small><sub>2</sub></small>. When a tetrapyridyl porphyrin analogue is post-synthetically tethered through a oxalate-bridged Rh<small><sub>2</sub></small> molecular clip, the selectivity improves further to 7.2% H<small><sub>2</sub></small>O<small><sub>2</sub></small>. In contrast, when a longer bis-hydroxybenzoquinato bridge is used in the clip, the selectivity drops back to 14.5% H<small><sub>2</sub></small>O<small><sub>2</sub></small>. Our most selective system also shows the highest current response and therefore our post-synthetic modification provides a kinetic enhancement as well. These results establish that straightforward coordination chemistry can be used for post-synthetic tuning of molecular ORR catalysts and when proper molecular clips are selected, marked enhancements to both selectivity and activity may be realized.</p>","PeriodicalId":79,"journal":{"name":"Inorganic Chemistry Frontiers","volume":" 17","pages":" 5557-5565"},"PeriodicalIF":6.4000,"publicationDate":"2024-07-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2024/qi/d4qi01219d?page=search","citationCount":"0","resultStr":"{\"title\":\"Post-synthetic modification of bis-iron(iii)-μ-oxo-porphyrin prisms to enhance oxygen reduction electrocatalysis†\",\"authors\":\"Daoyang Zhang, Lauren E. Rosch, Matthew R. Crawley and Timothy R. Cook\",\"doi\":\"10.1039/D4QI01219D\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Bis-iron(<small>III</small>)-μ-oxo porphyrins are known electrocatalysts for Oxygen Reduction Reaction (ORR) that favor four-electron four-proton chemistry. The facile formation of the μ-oxo motif makes forming cofacial structures straightforward unlike for other metalloporphyrins where the face-to-face geometry must be enforced by tethering groups. We have pioneered the use of coordination chemistry to obtain cofacial porphyrin ORR catalysts containing Co(<small>II</small>) ions. Here, we adapt our use of molecular clips for the post-synthetic modification of μ-oxo porphyrin dimers rather than as self-assembly building blocks. Although the cofacial geometry is inherent to the μ-oxo core, under ORR catalysis conditions, the dimer is rapidly cleaved, resulting in reactivity differences between traditional unclipped bis-iron(<small>III</small>)-μ-oxo porphyrins and our post-synthetically tethered architectures. We demonstrate our approach using two molecular clips that differ in length. The bis-iron(<small>III</small>)-μ-oxo precatalysts were characterized by <small><sup>1</sup></small>H NMR, ESI-MS, and molecular modeling to support the formation of cofacial prisms. Catalytic activity was studied electrochemically using cyclic voltammetry and hydrodynamic voltammetry. Whereas untethered bis-iron(<small>III</small>)-μ-oxo porphyrins are limited to heterogenous catalysis so that the dimeric structure is not lost upon the removal of the oxo bridge, our clipped architectures show significant catalytic current response under homogeneous conditions. Furthermore, when a shorter molecular clip is used as a post-synthetic tether, the selectivity under heterogeneous conditions is significantly enhanced: monomeric Fe(<small>III</small>) tetraphenylporphyrin (TPhP) generates 64.3% H<small><sub>2</sub></small>O<small><sub>2</sub></small>. When this same porphyrin is templated into a cofacial environment (Fe<small><sub>2</sub></small>O TPhP) the selectivity improves to 15.8% H<small><sub>2</sub></small>O<small><sub>2</sub></small>. When a tetrapyridyl porphyrin analogue is post-synthetically tethered through a oxalate-bridged Rh<small><sub>2</sub></small> molecular clip, the selectivity improves further to 7.2% H<small><sub>2</sub></small>O<small><sub>2</sub></small>. In contrast, when a longer bis-hydroxybenzoquinato bridge is used in the clip, the selectivity drops back to 14.5% H<small><sub>2</sub></small>O<small><sub>2</sub></small>. Our most selective system also shows the highest current response and therefore our post-synthetic modification provides a kinetic enhancement as well. These results establish that straightforward coordination chemistry can be used for post-synthetic tuning of molecular ORR catalysts and when proper molecular clips are selected, marked enhancements to both selectivity and activity may be realized.</p>\",\"PeriodicalId\":79,\"journal\":{\"name\":\"Inorganic Chemistry Frontiers\",\"volume\":\" 17\",\"pages\":\" 5557-5565\"},\"PeriodicalIF\":6.4000,\"publicationDate\":\"2024-07-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.rsc.org/en/content/articlepdf/2024/qi/d4qi01219d?page=search\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Inorganic Chemistry Frontiers\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2024/qi/d4qi01219d\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, INORGANIC & NUCLEAR\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Inorganic Chemistry Frontiers","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/qi/d4qi01219d","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, INORGANIC & NUCLEAR","Score":null,"Total":0}
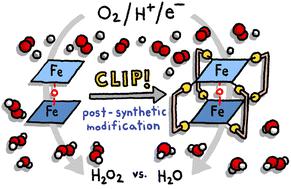
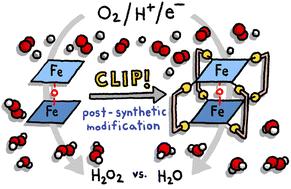
Post-synthetic modification of bis-iron(iii)-μ-oxo-porphyrin prisms to enhance oxygen reduction electrocatalysis†
Bis-iron(III)-μ-oxo porphyrins are known electrocatalysts for Oxygen Reduction Reaction (ORR) that favor four-electron four-proton chemistry. The facile formation of the μ-oxo motif makes forming cofacial structures straightforward unlike for other metalloporphyrins where the face-to-face geometry must be enforced by tethering groups. We have pioneered the use of coordination chemistry to obtain cofacial porphyrin ORR catalysts containing Co(II) ions. Here, we adapt our use of molecular clips for the post-synthetic modification of μ-oxo porphyrin dimers rather than as self-assembly building blocks. Although the cofacial geometry is inherent to the μ-oxo core, under ORR catalysis conditions, the dimer is rapidly cleaved, resulting in reactivity differences between traditional unclipped bis-iron(III)-μ-oxo porphyrins and our post-synthetically tethered architectures. We demonstrate our approach using two molecular clips that differ in length. The bis-iron(III)-μ-oxo precatalysts were characterized by 1H NMR, ESI-MS, and molecular modeling to support the formation of cofacial prisms. Catalytic activity was studied electrochemically using cyclic voltammetry and hydrodynamic voltammetry. Whereas untethered bis-iron(III)-μ-oxo porphyrins are limited to heterogenous catalysis so that the dimeric structure is not lost upon the removal of the oxo bridge, our clipped architectures show significant catalytic current response under homogeneous conditions. Furthermore, when a shorter molecular clip is used as a post-synthetic tether, the selectivity under heterogeneous conditions is significantly enhanced: monomeric Fe(III) tetraphenylporphyrin (TPhP) generates 64.3% H2O2. When this same porphyrin is templated into a cofacial environment (Fe2O TPhP) the selectivity improves to 15.8% H2O2. When a tetrapyridyl porphyrin analogue is post-synthetically tethered through a oxalate-bridged Rh2 molecular clip, the selectivity improves further to 7.2% H2O2. In contrast, when a longer bis-hydroxybenzoquinato bridge is used in the clip, the selectivity drops back to 14.5% H2O2. Our most selective system also shows the highest current response and therefore our post-synthetic modification provides a kinetic enhancement as well. These results establish that straightforward coordination chemistry can be used for post-synthetic tuning of molecular ORR catalysts and when proper molecular clips are selected, marked enhancements to both selectivity and activity may be realized.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


