Toni Vuković, Li Eon Kuek, Barbara Yu, Georgios Makris, Johannes Häberle
{"title":"枸橼酸缺乏症的治疗前景。","authors":"Toni Vuković, Li Eon Kuek, Barbara Yu, Georgios Makris, Johannes Häberle","doi":"10.1002/jimd.12768","DOIUrl":null,"url":null,"abstract":"<p>Citrin deficiency (CD) is a recessive, liver disease caused by sequence variants in the <i>SLC25A13</i> gene encoding a mitochondrial aspartate–glutamate transporter. CD manifests as different age-dependent phenotypes and affects crucial hepatic metabolic pathways including malate–aspartate-shuttle, glycolysis, gluconeogenesis, de novo lipogenesis and the tricarboxylic acid and urea cycles. Although the exact pathophysiology of CD remains unclear, impaired use of glucose and fatty acids as energy sources due to NADH shuttle defects and PPARα downregulation, respectively, indicates evident energy deficit in CD hepatocytes. The present review summarizes current trends on available and potential treatments for CD. Baseline recommendation for CD patients is dietary management, often already present as a self-selected food preference, that includes protein and fat-rich food, and avoidance of excess carbohydrates. At present, liver transplantation remains the sole curative option for severe CD cases. Our extensive literature review indicated medium-chain triglycerides (MCT) as the most widely used CD treatment in all age groups. MCT can effectively improve symptoms across disease phenotypes by rapidly supplying energy to the liver, restoring redox balance and inducing lipogenesis. In contrast, sodium pyruvate restored glycolysis and displayed initial preclinical promise, with however limited efficacy in adult CD patients. Ursodeoxycholic acid, nitrogen scavengers and <span>L</span>-arginine treatments effectively address specific pathophysiological aspects such as cholestasis and hyperammonemia and are commonly administered in combination with other drugs. Finally, future possibilities including restoring redox balance, amino acid supplementation, enhancing bioenergetics, improving ureagenesis and mRNA/DNA-based gene therapy are also discussed.</p>","PeriodicalId":16281,"journal":{"name":"Journal of Inherited Metabolic Disease","volume":"47 6","pages":"1157-1174"},"PeriodicalIF":4.2000,"publicationDate":"2024-07-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jimd.12768","citationCount":"0","resultStr":"{\"title\":\"The therapeutic landscape of citrin deficiency\",\"authors\":\"Toni Vuković, Li Eon Kuek, Barbara Yu, Georgios Makris, Johannes Häberle\",\"doi\":\"10.1002/jimd.12768\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Citrin deficiency (CD) is a recessive, liver disease caused by sequence variants in the <i>SLC25A13</i> gene encoding a mitochondrial aspartate–glutamate transporter. CD manifests as different age-dependent phenotypes and affects crucial hepatic metabolic pathways including malate–aspartate-shuttle, glycolysis, gluconeogenesis, de novo lipogenesis and the tricarboxylic acid and urea cycles. Although the exact pathophysiology of CD remains unclear, impaired use of glucose and fatty acids as energy sources due to NADH shuttle defects and PPARα downregulation, respectively, indicates evident energy deficit in CD hepatocytes. The present review summarizes current trends on available and potential treatments for CD. Baseline recommendation for CD patients is dietary management, often already present as a self-selected food preference, that includes protein and fat-rich food, and avoidance of excess carbohydrates. At present, liver transplantation remains the sole curative option for severe CD cases. Our extensive literature review indicated medium-chain triglycerides (MCT) as the most widely used CD treatment in all age groups. MCT can effectively improve symptoms across disease phenotypes by rapidly supplying energy to the liver, restoring redox balance and inducing lipogenesis. In contrast, sodium pyruvate restored glycolysis and displayed initial preclinical promise, with however limited efficacy in adult CD patients. Ursodeoxycholic acid, nitrogen scavengers and <span>L</span>-arginine treatments effectively address specific pathophysiological aspects such as cholestasis and hyperammonemia and are commonly administered in combination with other drugs. Finally, future possibilities including restoring redox balance, amino acid supplementation, enhancing bioenergetics, improving ureagenesis and mRNA/DNA-based gene therapy are also discussed.</p>\",\"PeriodicalId\":16281,\"journal\":{\"name\":\"Journal of Inherited Metabolic Disease\",\"volume\":\"47 6\",\"pages\":\"1157-1174\"},\"PeriodicalIF\":4.2000,\"publicationDate\":\"2024-07-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jimd.12768\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Inherited Metabolic Disease\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jimd.12768\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Inherited Metabolic Disease","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jimd.12768","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
摘要
枸橼酸苷缺乏症(CD)是一种隐性肝病,由编码线粒体天冬氨酸-谷氨酸转运体的 SLC25A13 基因的序列变异引起。CD 表现为不同年龄的表型,影响重要的肝脏代谢途径,包括苹果酸-天门冬氨酸-谷氨酸转运体、糖酵解、葡萄糖生成、新脂肪生成以及三羧酸循环和尿素循环。虽然 CD 的确切病理生理学仍不清楚,但由于 NADH 穿梭缺陷和 PPARα 下调,葡萄糖和脂肪酸作为能量来源的使用受损,这表明 CD 肝细胞明显能量不足。本综述总结了 CD 现有和潜在治疗方法的当前趋势。对 CD 患者的基本建议是饮食管理,通常已表现为自我选择食物偏好,包括蛋白质和脂肪丰富的食物,并避免摄入过多碳水化合物。目前,肝移植仍是治疗严重 CD 病例的唯一方法。我们广泛的文献综述表明,中链甘油三酯(MCT)是在所有年龄组中应用最广泛的 CD 治疗方法。中链甘油三酯可迅速为肝脏提供能量、恢复氧化还原平衡并诱导脂肪生成,从而有效改善各种疾病表型的症状。相比之下,丙酮酸钠可恢复糖酵解,并显示出初步的临床前前景,但对成年 CD 患者的疗效有限。熊去氧胆酸、氮清除剂和左旋精氨酸疗法可有效解决胆汁淤积和高氨血症等特定病理生理问题,通常与其他药物联合使用。最后,还讨论了未来的可能性,包括恢复氧化还原平衡、补充氨基酸、增强生物能、改善尿生成和基于 mRNA/DNA 的基因治疗。
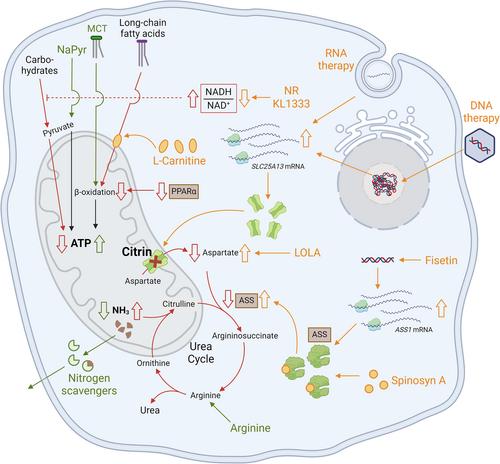
Citrin deficiency (CD) is a recessive, liver disease caused by sequence variants in the SLC25A13 gene encoding a mitochondrial aspartate–glutamate transporter. CD manifests as different age-dependent phenotypes and affects crucial hepatic metabolic pathways including malate–aspartate-shuttle, glycolysis, gluconeogenesis, de novo lipogenesis and the tricarboxylic acid and urea cycles. Although the exact pathophysiology of CD remains unclear, impaired use of glucose and fatty acids as energy sources due to NADH shuttle defects and PPARα downregulation, respectively, indicates evident energy deficit in CD hepatocytes. The present review summarizes current trends on available and potential treatments for CD. Baseline recommendation for CD patients is dietary management, often already present as a self-selected food preference, that includes protein and fat-rich food, and avoidance of excess carbohydrates. At present, liver transplantation remains the sole curative option for severe CD cases. Our extensive literature review indicated medium-chain triglycerides (MCT) as the most widely used CD treatment in all age groups. MCT can effectively improve symptoms across disease phenotypes by rapidly supplying energy to the liver, restoring redox balance and inducing lipogenesis. In contrast, sodium pyruvate restored glycolysis and displayed initial preclinical promise, with however limited efficacy in adult CD patients. Ursodeoxycholic acid, nitrogen scavengers and L-arginine treatments effectively address specific pathophysiological aspects such as cholestasis and hyperammonemia and are commonly administered in combination with other drugs. Finally, future possibilities including restoring redox balance, amino acid supplementation, enhancing bioenergetics, improving ureagenesis and mRNA/DNA-based gene therapy are also discussed.
期刊介绍:
The Journal of Inherited Metabolic Disease (JIMD) is the official journal of the Society for the Study of Inborn Errors of Metabolism (SSIEM). By enhancing communication between workers in the field throughout the world, the JIMD aims to improve the management and understanding of inherited metabolic disorders. It publishes results of original research and new or important observations pertaining to any aspect of inherited metabolic disease in humans and higher animals. This includes clinical (medical, dental and veterinary), biochemical, genetic (including cytogenetic, molecular and population genetic), experimental (including cell biological), methodological, theoretical, epidemiological, ethical and counselling aspects. The JIMD also reviews important new developments or controversial issues relating to metabolic disorders and publishes reviews and short reports arising from the Society''s annual symposia. A distinction is made between peer-reviewed scientific material that is selected because of its significance for other professionals in the field and non-peer- reviewed material that aims to be important, controversial, interesting or entertaining (“Extras”).

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


