Tomáš Seeman, Terezie Šuláková, Alice Bosáková, Jana Indráková, Dagmar Grečmalová
{"title":"首例 IFT140 杂合子缺失导致常染色体显性多囊肾的儿科病例:病例报告。","authors":"Tomáš Seeman, Terezie Šuláková, Alice Bosáková, Jana Indráková, Dagmar Grečmalová","doi":"10.1159/000539176","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Autosomal dominant polycystic kidney disease (ADPKD) is the most common hereditary kidney disease, which is mainly caused by pathogenic variants in two particular genes: <i>PKD1</i> and <i>PKD2</i>. ADPKD caused by variants in other genes (<i>GANAB</i> or <i>IFT140</i>) is very rare.</p><p><strong>Case report: </strong>In a 6-year-old girl examined for abdominal pain, a cystic mass in the upper part of the right kidney was detected during an abdominal ultrasound. She was referred to pediatric oncology and urology for suspicion of a tumorous mass and the condition was assessed as a cystic nephroma. A heminephrectomy was then performed on the upper cystic part of the right kidney. The histological examination was inconclusive; therefore, genetic testing was recommended. Kidney and liver cysts were detected sonographically in the mother, but DNA analysis of the <i>PKD1</i> and <i>PKD2</i> genes did not reveal any pathogenic variant; the cause of the pathological formation in the kidneys remained unclear. Nine years later, next-generation sequencing of a panel of genes for kidney disease was performed and a heterozygous deletion was found on chromosome 16; this included exon 13 of the <i>IFT140</i> gene. The same deletion was found in the patient's mother. Currently, the patient is 14 years old and has mild sonographic findings, normal glomerular filtration, mild proteinuria, and hypertension.</p><p><strong>Conclusion: </strong>Pathogenic variants of the <i>IFT140</i> gene very rarely cause ADPKD; however, they should be considered in all children with autosomal dominant forms of PKD and asymmetric/atypical cystic kidney involvement or negative findings of <i>PKD1</i> and <i>PKD2</i>.</p>","PeriodicalId":9599,"journal":{"name":"Case Reports in Nephrology and Dialysis","volume":"14 1","pages":"104-109"},"PeriodicalIF":0.9000,"publicationDate":"2024-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11249641/pdf/","citationCount":"0","resultStr":"{\"title\":\"The First Pediatric Case of an IFT140 Heterozygous Deletion Causing Autosomal Dominant Polycystic Kidney Disease: Case Report.\",\"authors\":\"Tomáš Seeman, Terezie Šuláková, Alice Bosáková, Jana Indráková, Dagmar Grečmalová\",\"doi\":\"10.1159/000539176\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Introduction: </strong>Autosomal dominant polycystic kidney disease (ADPKD) is the most common hereditary kidney disease, which is mainly caused by pathogenic variants in two particular genes: <i>PKD1</i> and <i>PKD2</i>. ADPKD caused by variants in other genes (<i>GANAB</i> or <i>IFT140</i>) is very rare.</p><p><strong>Case report: </strong>In a 6-year-old girl examined for abdominal pain, a cystic mass in the upper part of the right kidney was detected during an abdominal ultrasound. She was referred to pediatric oncology and urology for suspicion of a tumorous mass and the condition was assessed as a cystic nephroma. A heminephrectomy was then performed on the upper cystic part of the right kidney. The histological examination was inconclusive; therefore, genetic testing was recommended. Kidney and liver cysts were detected sonographically in the mother, but DNA analysis of the <i>PKD1</i> and <i>PKD2</i> genes did not reveal any pathogenic variant; the cause of the pathological formation in the kidneys remained unclear. Nine years later, next-generation sequencing of a panel of genes for kidney disease was performed and a heterozygous deletion was found on chromosome 16; this included exon 13 of the <i>IFT140</i> gene. The same deletion was found in the patient's mother. Currently, the patient is 14 years old and has mild sonographic findings, normal glomerular filtration, mild proteinuria, and hypertension.</p><p><strong>Conclusion: </strong>Pathogenic variants of the <i>IFT140</i> gene very rarely cause ADPKD; however, they should be considered in all children with autosomal dominant forms of PKD and asymmetric/atypical cystic kidney involvement or negative findings of <i>PKD1</i> and <i>PKD2</i>.</p>\",\"PeriodicalId\":9599,\"journal\":{\"name\":\"Case Reports in Nephrology and Dialysis\",\"volume\":\"14 1\",\"pages\":\"104-109\"},\"PeriodicalIF\":0.9000,\"publicationDate\":\"2024-07-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11249641/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Case Reports in Nephrology and Dialysis\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1159/000539176\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q4\",\"JCRName\":\"UROLOGY & NEPHROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Nephrology and Dialysis","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1159/000539176","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"UROLOGY & NEPHROLOGY","Score":null,"Total":0}
The First Pediatric Case of an IFT140 Heterozygous Deletion Causing Autosomal Dominant Polycystic Kidney Disease: Case Report.
Introduction: Autosomal dominant polycystic kidney disease (ADPKD) is the most common hereditary kidney disease, which is mainly caused by pathogenic variants in two particular genes: PKD1 and PKD2. ADPKD caused by variants in other genes (GANAB or IFT140) is very rare.
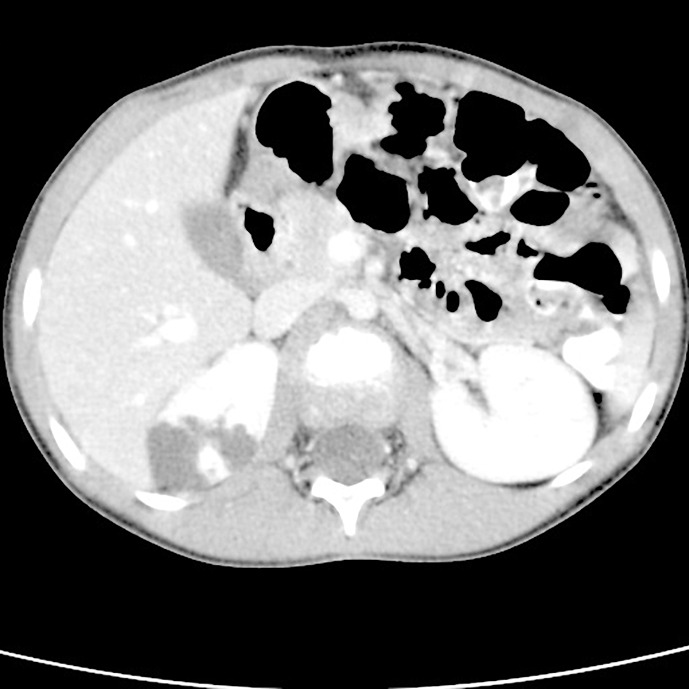
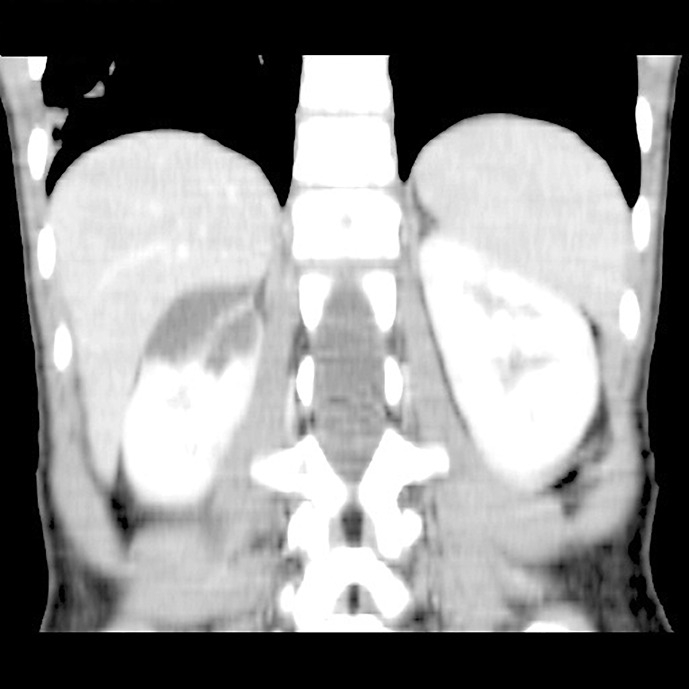
Case report: In a 6-year-old girl examined for abdominal pain, a cystic mass in the upper part of the right kidney was detected during an abdominal ultrasound. She was referred to pediatric oncology and urology for suspicion of a tumorous mass and the condition was assessed as a cystic nephroma. A heminephrectomy was then performed on the upper cystic part of the right kidney. The histological examination was inconclusive; therefore, genetic testing was recommended. Kidney and liver cysts were detected sonographically in the mother, but DNA analysis of the PKD1 and PKD2 genes did not reveal any pathogenic variant; the cause of the pathological formation in the kidneys remained unclear. Nine years later, next-generation sequencing of a panel of genes for kidney disease was performed and a heterozygous deletion was found on chromosome 16; this included exon 13 of the IFT140 gene. The same deletion was found in the patient's mother. Currently, the patient is 14 years old and has mild sonographic findings, normal glomerular filtration, mild proteinuria, and hypertension.
Conclusion: Pathogenic variants of the IFT140 gene very rarely cause ADPKD; however, they should be considered in all children with autosomal dominant forms of PKD and asymmetric/atypical cystic kidney involvement or negative findings of PKD1 and PKD2.
期刊介绍:
This peer-reviewed online-only journal publishes original case reports covering the entire spectrum of nephrology and dialysis, including genetic susceptibility, clinical presentation, diagnosis, treatment or prevention, toxicities of therapy, critical care, supportive care, quality-of-life and survival issues. The journal will also accept case reports dealing with the use of novel technologies, both in the arena of diagnosis and treatment. Supplementary material is welcomed.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


