Puck van Gerwen, Ksenia R Briling, Charlotte Bunne, Vignesh Ram Somnath, Ruben Laplaza, Andreas Krause, Clemence Corminboeuf
{"title":"3DReact:化学反应几何深度学习。","authors":"Puck van Gerwen, Ksenia R Briling, Charlotte Bunne, Vignesh Ram Somnath, Ruben Laplaza, Andreas Krause, Clemence Corminboeuf","doi":"10.1021/acs.jcim.4c00104","DOIUrl":null,"url":null,"abstract":"<p><p>Geometric deep learning models, which incorporate the relevant molecular symmetries within the neural network architecture, have considerably improved the accuracy and data efficiency of predictions of molecular properties. Building on this success, we introduce 3DReact, a geometric deep learning model to predict reaction properties from three-dimensional structures of reactants and products. We demonstrate that the invariant version of the model is sufficient for existing reaction data sets. We illustrate its competitive performance on the prediction of activation barriers on the GDB7-22-TS, Cyclo-23-TS, and Proparg-21-TS data sets in different atom-mapping regimes. We show that, compared to existing models for reaction property prediction, 3DReact offers a flexible framework that exploits atom-mapping information, if available, as well as geometries of reactants and products (in an invariant or equivariant fashion). Accordingly, it performs systematically well across different data sets, atom-mapping regimes, as well as both interpolation and extrapolation tasks.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":" ","pages":"5771-5785"},"PeriodicalIF":5.3000,"publicationDate":"2024-08-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11323278/pdf/","citationCount":"0","resultStr":"{\"title\":\"3DReact: Geometric Deep Learning for Chemical Reactions.\",\"authors\":\"Puck van Gerwen, Ksenia R Briling, Charlotte Bunne, Vignesh Ram Somnath, Ruben Laplaza, Andreas Krause, Clemence Corminboeuf\",\"doi\":\"10.1021/acs.jcim.4c00104\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Geometric deep learning models, which incorporate the relevant molecular symmetries within the neural network architecture, have considerably improved the accuracy and data efficiency of predictions of molecular properties. Building on this success, we introduce 3DReact, a geometric deep learning model to predict reaction properties from three-dimensional structures of reactants and products. We demonstrate that the invariant version of the model is sufficient for existing reaction data sets. We illustrate its competitive performance on the prediction of activation barriers on the GDB7-22-TS, Cyclo-23-TS, and Proparg-21-TS data sets in different atom-mapping regimes. We show that, compared to existing models for reaction property prediction, 3DReact offers a flexible framework that exploits atom-mapping information, if available, as well as geometries of reactants and products (in an invariant or equivariant fashion). Accordingly, it performs systematically well across different data sets, atom-mapping regimes, as well as both interpolation and extrapolation tasks.</p>\",\"PeriodicalId\":44,\"journal\":{\"name\":\"Journal of Chemical Information and Modeling \",\"volume\":\" \",\"pages\":\"5771-5785\"},\"PeriodicalIF\":5.3000,\"publicationDate\":\"2024-08-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11323278/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Information and Modeling \",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jcim.4c00104\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/7/15 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jcim.4c00104","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/15 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
3DReact: Geometric Deep Learning for Chemical Reactions.
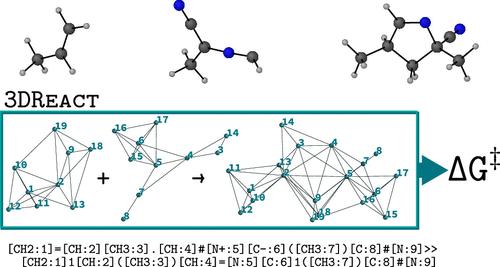
Geometric deep learning models, which incorporate the relevant molecular symmetries within the neural network architecture, have considerably improved the accuracy and data efficiency of predictions of molecular properties. Building on this success, we introduce 3DReact, a geometric deep learning model to predict reaction properties from three-dimensional structures of reactants and products. We demonstrate that the invariant version of the model is sufficient for existing reaction data sets. We illustrate its competitive performance on the prediction of activation barriers on the GDB7-22-TS, Cyclo-23-TS, and Proparg-21-TS data sets in different atom-mapping regimes. We show that, compared to existing models for reaction property prediction, 3DReact offers a flexible framework that exploits atom-mapping information, if available, as well as geometries of reactants and products (in an invariant or equivariant fashion). Accordingly, it performs systematically well across different data sets, atom-mapping regimes, as well as both interpolation and extrapolation tasks.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


