{"title":"枸橼酸缺乏症--东边的故事。","authors":"Johannes Häberle","doi":"10.1002/jimd.12772","DOIUrl":null,"url":null,"abstract":"<p>Citrin deficiency (CD) is a complex metabolic condition due to defects in <i>SLC25A13</i> encoding citrin, an aspartate/glutamate carrier located in the mitochondrial inner membrane. The condition was first described in Japan and other East Asian countries in patients who were thought to suffer from classical citrullinemia type 1, and was therefore classified as a urea cycle disorder. With an improved understanding of its molecular basis, it became apparent that a defect of citrin is primarily affecting the malate–aspartate shuttle with however multiple secondary effects on many central metabolic pathways including glycolysis, gluconeogenesis, de novo lipogenesis and ureagenesis. In the meantime, it became also clear that CD must be considered as a global disease with patients identified in many parts of the world and affected by <i>SLC25A13</i> genotypes different from those known in East Asian populations. The present short review summarizes the (hi)story of this complex metabolic condition and tries to explain the relevance of including CD as a differential diagnosis in neonates and infants with cholestasis and in (not only adult) patients with hyperammonemia of unknown origin with subsequent impact on the emergency management.</p>","PeriodicalId":16281,"journal":{"name":"Journal of Inherited Metabolic Disease","volume":"47 6","pages":"1129-1133"},"PeriodicalIF":4.2000,"publicationDate":"2024-07-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jimd.12772","citationCount":"0","resultStr":"{\"title\":\"Citrin deficiency—The East-side story\",\"authors\":\"Johannes Häberle\",\"doi\":\"10.1002/jimd.12772\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Citrin deficiency (CD) is a complex metabolic condition due to defects in <i>SLC25A13</i> encoding citrin, an aspartate/glutamate carrier located in the mitochondrial inner membrane. The condition was first described in Japan and other East Asian countries in patients who were thought to suffer from classical citrullinemia type 1, and was therefore classified as a urea cycle disorder. With an improved understanding of its molecular basis, it became apparent that a defect of citrin is primarily affecting the malate–aspartate shuttle with however multiple secondary effects on many central metabolic pathways including glycolysis, gluconeogenesis, de novo lipogenesis and ureagenesis. In the meantime, it became also clear that CD must be considered as a global disease with patients identified in many parts of the world and affected by <i>SLC25A13</i> genotypes different from those known in East Asian populations. The present short review summarizes the (hi)story of this complex metabolic condition and tries to explain the relevance of including CD as a differential diagnosis in neonates and infants with cholestasis and in (not only adult) patients with hyperammonemia of unknown origin with subsequent impact on the emergency management.</p>\",\"PeriodicalId\":16281,\"journal\":{\"name\":\"Journal of Inherited Metabolic Disease\",\"volume\":\"47 6\",\"pages\":\"1129-1133\"},\"PeriodicalIF\":4.2000,\"publicationDate\":\"2024-07-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jimd.12772\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Inherited Metabolic Disease\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jimd.12772\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Inherited Metabolic Disease","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jimd.12772","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
摘要
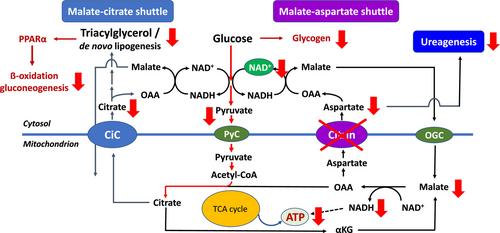
柠檬蛋白缺乏症(CD)是一种复杂的代谢性疾病,由于编码柠檬蛋白的 SLC25A13 存在缺陷,而柠檬蛋白是一种位于线粒体内膜的天冬氨酸/谷氨酸载体。该病最早出现在日本和其他东亚国家,患者被认为患有典型的瓜氨酸血症 1 型,因此被归类为尿素循环障碍。随着对其分子基础的进一步了解,人们发现柠檬素的缺陷主要影响苹果酸-天门冬氨酸穿梭,但对许多中枢代谢途径(包括糖酵解、葡萄糖生成、新生脂肪生成和尿素生成)具有多重继发性影响。与此同时,人们还清楚地认识到,CD 必须被视为一种全球性疾病,在世界许多地方都能发现 CD 患者,他们的 SLC25A13 基因型与东亚人群中已知的基因型不同。本短文总结了这一复杂代谢疾病的(历史)故事,并试图解释将 CD 作为胆汁淤积症新生儿和婴儿以及不明原因高氨血症(不仅是成人)患者的鉴别诊断的相关性,以及随后对紧急处理的影响。
Citrin deficiency (CD) is a complex metabolic condition due to defects in SLC25A13 encoding citrin, an aspartate/glutamate carrier located in the mitochondrial inner membrane. The condition was first described in Japan and other East Asian countries in patients who were thought to suffer from classical citrullinemia type 1, and was therefore classified as a urea cycle disorder. With an improved understanding of its molecular basis, it became apparent that a defect of citrin is primarily affecting the malate–aspartate shuttle with however multiple secondary effects on many central metabolic pathways including glycolysis, gluconeogenesis, de novo lipogenesis and ureagenesis. In the meantime, it became also clear that CD must be considered as a global disease with patients identified in many parts of the world and affected by SLC25A13 genotypes different from those known in East Asian populations. The present short review summarizes the (hi)story of this complex metabolic condition and tries to explain the relevance of including CD as a differential diagnosis in neonates and infants with cholestasis and in (not only adult) patients with hyperammonemia of unknown origin with subsequent impact on the emergency management.
期刊介绍:
The Journal of Inherited Metabolic Disease (JIMD) is the official journal of the Society for the Study of Inborn Errors of Metabolism (SSIEM). By enhancing communication between workers in the field throughout the world, the JIMD aims to improve the management and understanding of inherited metabolic disorders. It publishes results of original research and new or important observations pertaining to any aspect of inherited metabolic disease in humans and higher animals. This includes clinical (medical, dental and veterinary), biochemical, genetic (including cytogenetic, molecular and population genetic), experimental (including cell biological), methodological, theoretical, epidemiological, ethical and counselling aspects. The JIMD also reviews important new developments or controversial issues relating to metabolic disorders and publishes reviews and short reports arising from the Society''s annual symposia. A distinction is made between peer-reviewed scientific material that is selected because of its significance for other professionals in the field and non-peer- reviewed material that aims to be important, controversial, interesting or entertaining (“Extras”).

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


