Francesca Peranzoni, Roberto De Castro, Emilio Merlini, Yen Le Nguyen
{"title":"46 XX卵巢性发育障碍伴促性腺激素释放激素受体、PROKR2基因常染色体隐性杂合子缺义突变和常染色体显性杂合子缺义突变:病例报告。","authors":"Francesca Peranzoni, Roberto De Castro, Emilio Merlini, Yen Le Nguyen","doi":"10.1055/s-0044-1788060","DOIUrl":null,"url":null,"abstract":"<p><p>True hermaphroditism is a disorder of sex development (DSD), accounting for less than 5% of all DSD cases, defined by the simultaneous presence of testicular tissue and ovarian tissue in the same individual. In the reported case, the patient presented two genetic mutations involved in the pathogenic pathway of the DSD condition associated with the clinical features of Kallmann syndrome (KS), a developmental disease that associates hypogonadotropic hypogonadism (HH), due to gonadotropin-releasing hormone deficiency, and anosmia, related to the absence or hypoplasia of the olfactory bulbs. Given the variable degree of hyposmia in KS, the distinction between KS and normosmic idiopathic HH is currently unclear, especially as HH patients do not always undergo detailed olfactory testing. This syndrome is very rare, with an estimated prevalence of 1:80,000 in males and 1:40,000 in females. This is the only case report concerning a patient with 46 XX true hermaphroditism affected by HH and digenic inheritance of Kallmann syndrome.</p>","PeriodicalId":40142,"journal":{"name":"Global Medical Genetics","volume":"11 3","pages":"220-224"},"PeriodicalIF":1.5000,"publicationDate":"2024-07-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11233268/pdf/","citationCount":"0","resultStr":"{\"title\":\"46 XX Ovotesticular Disorder of Sex Development with Gonadotropin-Releasing Hormone Receptor, Autosomal Recessive Heterozygous Missense Mutation and Autosomal Dominant Heterozygous Missense Mutation of the <i>PROKR2</i> Gene: A Case Report.\",\"authors\":\"Francesca Peranzoni, Roberto De Castro, Emilio Merlini, Yen Le Nguyen\",\"doi\":\"10.1055/s-0044-1788060\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>True hermaphroditism is a disorder of sex development (DSD), accounting for less than 5% of all DSD cases, defined by the simultaneous presence of testicular tissue and ovarian tissue in the same individual. In the reported case, the patient presented two genetic mutations involved in the pathogenic pathway of the DSD condition associated with the clinical features of Kallmann syndrome (KS), a developmental disease that associates hypogonadotropic hypogonadism (HH), due to gonadotropin-releasing hormone deficiency, and anosmia, related to the absence or hypoplasia of the olfactory bulbs. Given the variable degree of hyposmia in KS, the distinction between KS and normosmic idiopathic HH is currently unclear, especially as HH patients do not always undergo detailed olfactory testing. This syndrome is very rare, with an estimated prevalence of 1:80,000 in males and 1:40,000 in females. This is the only case report concerning a patient with 46 XX true hermaphroditism affected by HH and digenic inheritance of Kallmann syndrome.</p>\",\"PeriodicalId\":40142,\"journal\":{\"name\":\"Global Medical Genetics\",\"volume\":\"11 3\",\"pages\":\"220-224\"},\"PeriodicalIF\":1.5000,\"publicationDate\":\"2024-07-09\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11233268/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Global Medical Genetics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1055/s-0044-1788060\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/9/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q4\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Global Medical Genetics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1055/s-0044-1788060","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
46 XX Ovotesticular Disorder of Sex Development with Gonadotropin-Releasing Hormone Receptor, Autosomal Recessive Heterozygous Missense Mutation and Autosomal Dominant Heterozygous Missense Mutation of the PROKR2 Gene: A Case Report.
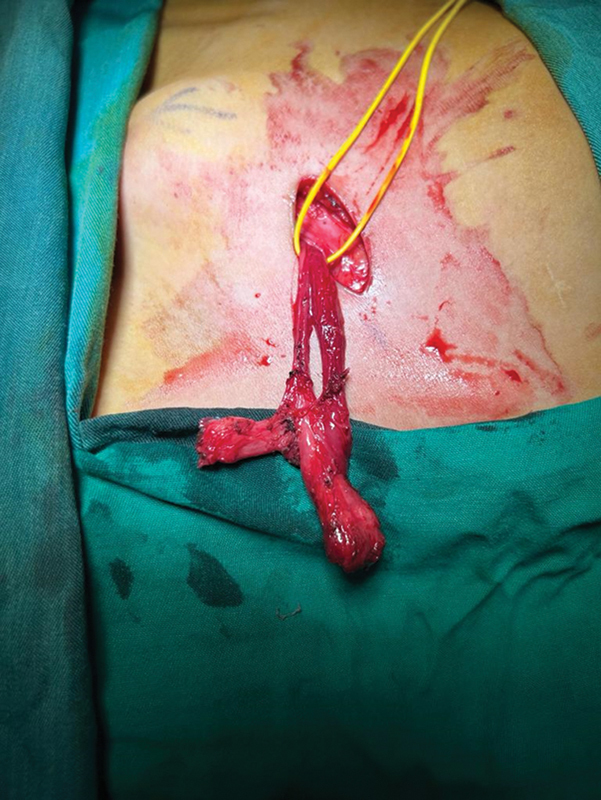
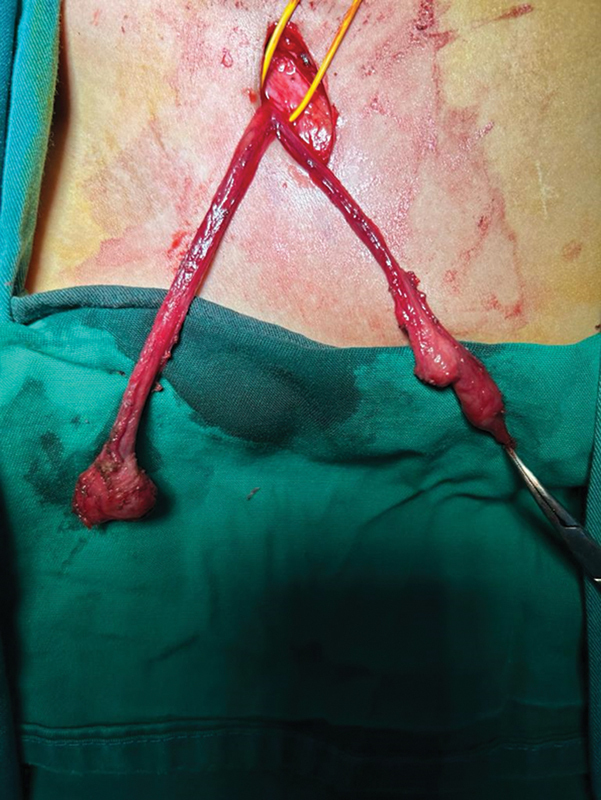
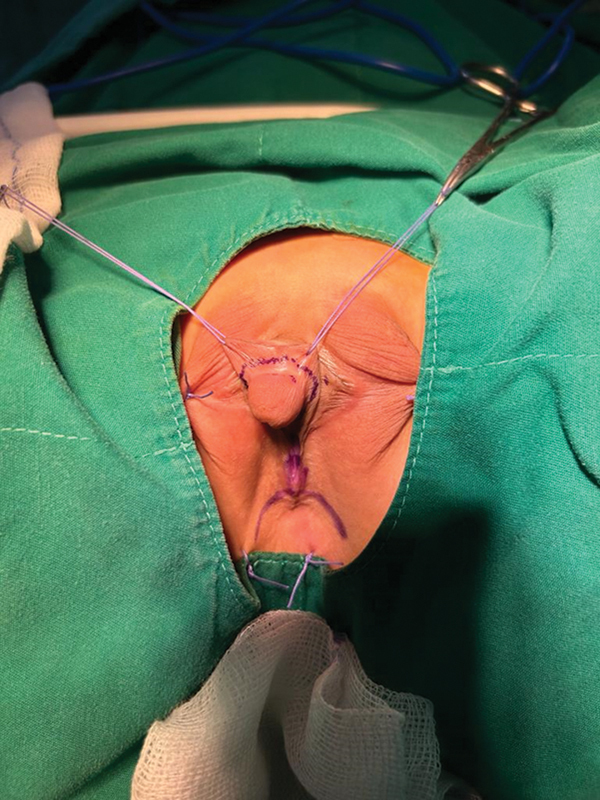
True hermaphroditism is a disorder of sex development (DSD), accounting for less than 5% of all DSD cases, defined by the simultaneous presence of testicular tissue and ovarian tissue in the same individual. In the reported case, the patient presented two genetic mutations involved in the pathogenic pathway of the DSD condition associated with the clinical features of Kallmann syndrome (KS), a developmental disease that associates hypogonadotropic hypogonadism (HH), due to gonadotropin-releasing hormone deficiency, and anosmia, related to the absence or hypoplasia of the olfactory bulbs. Given the variable degree of hyposmia in KS, the distinction between KS and normosmic idiopathic HH is currently unclear, especially as HH patients do not always undergo detailed olfactory testing. This syndrome is very rare, with an estimated prevalence of 1:80,000 in males and 1:40,000 in females. This is the only case report concerning a patient with 46 XX true hermaphroditism affected by HH and digenic inheritance of Kallmann syndrome.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


