{"title":"设计针对 SARS-CoV-2 Mpro 酶的新型抑制剂衍生物:一种深度学习和结构生物学方法","authors":"Tushar Joshi, Shalini Mathpal, Priyanka Sharma, Akshay Abraham, Rajadurai Vijay Solomon and Subhash Chandra","doi":"10.1039/D4ME00062E","DOIUrl":null,"url":null,"abstract":"<p >The emerging variants of SARS-CoV-2 have raised serious concerns worldwide due to their infectivity, lethality, and unpredictability. Moreover, the ability of these variants to bypass vaccine protection and immunity has compelled the research community to design novel compounds against SARS-CoV-2. This study focuses on designing novel molecules using artificial intelligence methods for the development of new therapeutics against SARS-CoV-2. Furthermore, these molecules were validated against main protease (M<small><sup>pro</sup></small>) using <em>in-silico</em> methods. In this study, we used the DeepScreening RNN-based web server to design novel molecules using potential inhibitors of M<small><sup>pro</sup></small> from CHEMBL4495582. Screened compounds were further validated by molecular docking and molecular dynamics (MD) simulation studies. One hundred molecules were obtained and studied through molecular docking and MD simulations. Additionally, eight molecules, based on their docking scores, were also evaluated for electronic structure properties by conducting Density Functional Theory (DFT) calculations using the B3LYP method and a 6-31G basis set. A total of three compounds, namely L18, L36, and L26, showed very good binding and stability with the active site of the M<small><sup>pro</sup></small> protein. The results of this study demonstrate that potential molecules can be designed using artificial intelligence methods for the rapid development of drug candidates against SARS-CoV-2, addressing the alarming worldwide situation of emerging deadly SARS-CoV-2 variants. We hope that our study will attract the attention of the scientific community to increase the application of artificial intelligence techniques in the drug discovery process.</p>","PeriodicalId":91,"journal":{"name":"Molecular Systems Design & Engineering","volume":" 10","pages":" 1063-1076"},"PeriodicalIF":3.2000,"publicationDate":"2024-07-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Designing novel inhibitor derivatives targeting SARS-CoV-2 Mpro enzyme: a deep learning and structure biology approach†\",\"authors\":\"Tushar Joshi, Shalini Mathpal, Priyanka Sharma, Akshay Abraham, Rajadurai Vijay Solomon and Subhash Chandra\",\"doi\":\"10.1039/D4ME00062E\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The emerging variants of SARS-CoV-2 have raised serious concerns worldwide due to their infectivity, lethality, and unpredictability. Moreover, the ability of these variants to bypass vaccine protection and immunity has compelled the research community to design novel compounds against SARS-CoV-2. This study focuses on designing novel molecules using artificial intelligence methods for the development of new therapeutics against SARS-CoV-2. Furthermore, these molecules were validated against main protease (M<small><sup>pro</sup></small>) using <em>in-silico</em> methods. In this study, we used the DeepScreening RNN-based web server to design novel molecules using potential inhibitors of M<small><sup>pro</sup></small> from CHEMBL4495582. Screened compounds were further validated by molecular docking and molecular dynamics (MD) simulation studies. One hundred molecules were obtained and studied through molecular docking and MD simulations. Additionally, eight molecules, based on their docking scores, were also evaluated for electronic structure properties by conducting Density Functional Theory (DFT) calculations using the B3LYP method and a 6-31G basis set. A total of three compounds, namely L18, L36, and L26, showed very good binding and stability with the active site of the M<small><sup>pro</sup></small> protein. The results of this study demonstrate that potential molecules can be designed using artificial intelligence methods for the rapid development of drug candidates against SARS-CoV-2, addressing the alarming worldwide situation of emerging deadly SARS-CoV-2 variants. We hope that our study will attract the attention of the scientific community to increase the application of artificial intelligence techniques in the drug discovery process.</p>\",\"PeriodicalId\":91,\"journal\":{\"name\":\"Molecular Systems Design & Engineering\",\"volume\":\" 10\",\"pages\":\" 1063-1076\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2024-07-10\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Systems Design & Engineering\",\"FirstCategoryId\":\"5\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2024/me/d4me00062e\",\"RegionNum\":3,\"RegionCategory\":\"工程技术\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Systems Design & Engineering","FirstCategoryId":"5","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/me/d4me00062e","RegionNum":3,"RegionCategory":"工程技术","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Designing novel inhibitor derivatives targeting SARS-CoV-2 Mpro enzyme: a deep learning and structure biology approach†
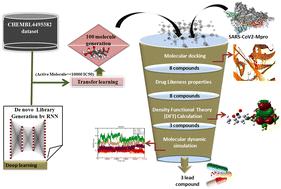
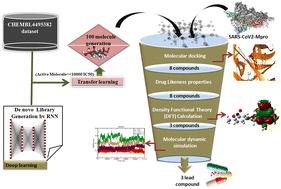
The emerging variants of SARS-CoV-2 have raised serious concerns worldwide due to their infectivity, lethality, and unpredictability. Moreover, the ability of these variants to bypass vaccine protection and immunity has compelled the research community to design novel compounds against SARS-CoV-2. This study focuses on designing novel molecules using artificial intelligence methods for the development of new therapeutics against SARS-CoV-2. Furthermore, these molecules were validated against main protease (Mpro) using in-silico methods. In this study, we used the DeepScreening RNN-based web server to design novel molecules using potential inhibitors of Mpro from CHEMBL4495582. Screened compounds were further validated by molecular docking and molecular dynamics (MD) simulation studies. One hundred molecules were obtained and studied through molecular docking and MD simulations. Additionally, eight molecules, based on their docking scores, were also evaluated for electronic structure properties by conducting Density Functional Theory (DFT) calculations using the B3LYP method and a 6-31G basis set. A total of three compounds, namely L18, L36, and L26, showed very good binding and stability with the active site of the Mpro protein. The results of this study demonstrate that potential molecules can be designed using artificial intelligence methods for the rapid development of drug candidates against SARS-CoV-2, addressing the alarming worldwide situation of emerging deadly SARS-CoV-2 variants. We hope that our study will attract the attention of the scientific community to increase the application of artificial intelligence techniques in the drug discovery process.
期刊介绍:
Molecular Systems Design & Engineering provides a hub for cutting-edge research into how understanding of molecular properties, behaviour and interactions can be used to design and assemble better materials, systems, and processes to achieve specific functions. These may have applications of technological significance and help address global challenges.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


