Guillermo Vazquez , Daniel Sauceda , Raymundo Arróyave
{"title":"解密高熵材料中的化学排序:机器学习加速的高通量集群扩展方法","authors":"Guillermo Vazquez , Daniel Sauceda , Raymundo Arróyave","doi":"10.1016/j.actamat.2024.120137","DOIUrl":null,"url":null,"abstract":"<div><p>The Cluster Expansion (CE) Method encounters significant computational challenges in multicomponent systems due to the computational expense of generating training data through density functional theory (DFT) calculations. This work aims to refine the cluster and structure selection processes to mitigate these challenges. We introduce a novel method that significantly reduces the computational load associated with the calculation of fitting parameters. This method employs a Graph Neural Network (GNN) model, leveraging the M3GNet network, which is trained using a select subset of DFT calculations at each ionic step. The trained surrogate model excels in predicting the volume and energy of the final structure for a relaxation run. By employing this model, we sample thousands of structures and fit a CE model to the energies of these GNN-relaxed structures. This approach, utilizing a large training dataset, effectively reduces the risk of overfitting, yielding a CE model with a root-mean-square error (RMSE) below 10 meV/atom. We validate our method’s effectiveness in two test cases: the (Ti, Cr, Zr, Mo, Hf, Ta)B<span><math><msub><mrow></mrow><mrow><mn>2</mn></mrow></msub></math></span> diboride system and the Refractory High-Entropy Alloy (HEA) AlTiZrNbHfTa system. Our findings demonstrate the significant advantages of integrating a GNN model, specifically the M3GNet network, with CE methods for the efficient predictive analysis of chemical ordering in High Entropy Materials. The accelerating capabilities of the hybrid ML-CE approach to investigate the evolution of Short Range Ordering (SRO) in a large number of stoichiometric systems. Finally, we show how it is possible to correlate the strength of chemical ordering to easily accessible alloy parameters.</p></div>","PeriodicalId":238,"journal":{"name":"Acta Materialia","volume":null,"pages":null},"PeriodicalIF":8.3000,"publicationDate":"2024-06-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Deciphering chemical ordering in High Entropy Materials: A machine learning-accelerated high-throughput cluster expansion approach\",\"authors\":\"Guillermo Vazquez , Daniel Sauceda , Raymundo Arróyave\",\"doi\":\"10.1016/j.actamat.2024.120137\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>The Cluster Expansion (CE) Method encounters significant computational challenges in multicomponent systems due to the computational expense of generating training data through density functional theory (DFT) calculations. This work aims to refine the cluster and structure selection processes to mitigate these challenges. We introduce a novel method that significantly reduces the computational load associated with the calculation of fitting parameters. This method employs a Graph Neural Network (GNN) model, leveraging the M3GNet network, which is trained using a select subset of DFT calculations at each ionic step. The trained surrogate model excels in predicting the volume and energy of the final structure for a relaxation run. By employing this model, we sample thousands of structures and fit a CE model to the energies of these GNN-relaxed structures. This approach, utilizing a large training dataset, effectively reduces the risk of overfitting, yielding a CE model with a root-mean-square error (RMSE) below 10 meV/atom. We validate our method’s effectiveness in two test cases: the (Ti, Cr, Zr, Mo, Hf, Ta)B<span><math><msub><mrow></mrow><mrow><mn>2</mn></mrow></msub></math></span> diboride system and the Refractory High-Entropy Alloy (HEA) AlTiZrNbHfTa system. Our findings demonstrate the significant advantages of integrating a GNN model, specifically the M3GNet network, with CE methods for the efficient predictive analysis of chemical ordering in High Entropy Materials. The accelerating capabilities of the hybrid ML-CE approach to investigate the evolution of Short Range Ordering (SRO) in a large number of stoichiometric systems. Finally, we show how it is possible to correlate the strength of chemical ordering to easily accessible alloy parameters.</p></div>\",\"PeriodicalId\":238,\"journal\":{\"name\":\"Acta Materialia\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":8.3000,\"publicationDate\":\"2024-06-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Acta Materialia\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S1359645424004889\",\"RegionNum\":1,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MATERIALS SCIENCE, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta Materialia","FirstCategoryId":"88","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1359645424004889","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATERIALS SCIENCE, MULTIDISCIPLINARY","Score":null,"Total":0}
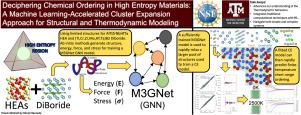
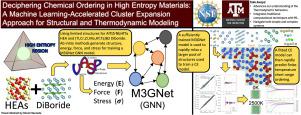
Deciphering chemical ordering in High Entropy Materials: A machine learning-accelerated high-throughput cluster expansion approach
The Cluster Expansion (CE) Method encounters significant computational challenges in multicomponent systems due to the computational expense of generating training data through density functional theory (DFT) calculations. This work aims to refine the cluster and structure selection processes to mitigate these challenges. We introduce a novel method that significantly reduces the computational load associated with the calculation of fitting parameters. This method employs a Graph Neural Network (GNN) model, leveraging the M3GNet network, which is trained using a select subset of DFT calculations at each ionic step. The trained surrogate model excels in predicting the volume and energy of the final structure for a relaxation run. By employing this model, we sample thousands of structures and fit a CE model to the energies of these GNN-relaxed structures. This approach, utilizing a large training dataset, effectively reduces the risk of overfitting, yielding a CE model with a root-mean-square error (RMSE) below 10 meV/atom. We validate our method’s effectiveness in two test cases: the (Ti, Cr, Zr, Mo, Hf, Ta)B diboride system and the Refractory High-Entropy Alloy (HEA) AlTiZrNbHfTa system. Our findings demonstrate the significant advantages of integrating a GNN model, specifically the M3GNet network, with CE methods for the efficient predictive analysis of chemical ordering in High Entropy Materials. The accelerating capabilities of the hybrid ML-CE approach to investigate the evolution of Short Range Ordering (SRO) in a large number of stoichiometric systems. Finally, we show how it is possible to correlate the strength of chemical ordering to easily accessible alloy parameters.
期刊介绍:
Acta Materialia serves as a platform for publishing full-length, original papers and commissioned overviews that contribute to a profound understanding of the correlation between the processing, structure, and properties of inorganic materials. The journal seeks papers with high impact potential or those that significantly propel the field forward. The scope includes the atomic and molecular arrangements, chemical and electronic structures, and microstructure of materials, focusing on their mechanical or functional behavior across all length scales, including nanostructures.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


