{"title":"确定以大肠杆菌核糖体解码中心为靶点的氨基糖苷类药物候选药物的计算工作流程。","authors":"Merve Yuce , Beril Ates , Nesrin Isil Yasar , Fethiye Aylin Sungur , Ozge Kurkcuoglu","doi":"10.1016/j.jmgm.2024.108817","DOIUrl":null,"url":null,"abstract":"<div><p>The global antibiotic resistance problem necessitates fast and effective approaches to finding novel inhibitors to treat bacterial infections. In this study, we propose a computational workflow to identify plausible high-affinity compounds from FDA-approved, investigational, and experimental libraries for the decoding center on the small subunit 30S of the <em>E. coli</em> ribosome. The workflow basically consists of two molecular docking calculations on the intact 30S, followed by molecular dynamics (MD) simulations coupled with MM-GBSA calculations on a truncated ribosome structure. The parameters used in the molecular docking suits, Glide and AutoDock Vina, as well as in the MD simulations with Desmond were carefully adjusted to obtain expected interactions for the ligand-rRNA complexes. A filtering procedure was followed, considering a fingerprint based on aminoglycoside's binding site on the 30S to obtain seven hit compounds either with different clinical usages or aminoglycoside derivatives under investigation, suggested for <em>in vitro</em> studies. The detailed workflow developed in this study promises an effective and fast approach for the estimation of binding free energies of large protein-RNA and ligand complexes.</p></div>","PeriodicalId":16361,"journal":{"name":"Journal of molecular graphics & modelling","volume":"131 ","pages":"Article 108817"},"PeriodicalIF":2.7000,"publicationDate":"2024-07-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"A computational workflow to determine drug candidates alternative to aminoglycosides targeting the decoding center of E. coli ribosome\",\"authors\":\"Merve Yuce , Beril Ates , Nesrin Isil Yasar , Fethiye Aylin Sungur , Ozge Kurkcuoglu\",\"doi\":\"10.1016/j.jmgm.2024.108817\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>The global antibiotic resistance problem necessitates fast and effective approaches to finding novel inhibitors to treat bacterial infections. In this study, we propose a computational workflow to identify plausible high-affinity compounds from FDA-approved, investigational, and experimental libraries for the decoding center on the small subunit 30S of the <em>E. coli</em> ribosome. The workflow basically consists of two molecular docking calculations on the intact 30S, followed by molecular dynamics (MD) simulations coupled with MM-GBSA calculations on a truncated ribosome structure. The parameters used in the molecular docking suits, Glide and AutoDock Vina, as well as in the MD simulations with Desmond were carefully adjusted to obtain expected interactions for the ligand-rRNA complexes. A filtering procedure was followed, considering a fingerprint based on aminoglycoside's binding site on the 30S to obtain seven hit compounds either with different clinical usages or aminoglycoside derivatives under investigation, suggested for <em>in vitro</em> studies. The detailed workflow developed in this study promises an effective and fast approach for the estimation of binding free energies of large protein-RNA and ligand complexes.</p></div>\",\"PeriodicalId\":16361,\"journal\":{\"name\":\"Journal of molecular graphics & modelling\",\"volume\":\"131 \",\"pages\":\"Article 108817\"},\"PeriodicalIF\":2.7000,\"publicationDate\":\"2024-07-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of molecular graphics & modelling\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S1093326324001177\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular graphics & modelling","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1093326324001177","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
A computational workflow to determine drug candidates alternative to aminoglycosides targeting the decoding center of E. coli ribosome
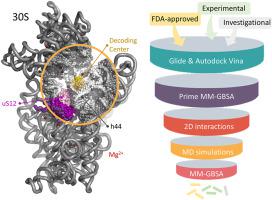
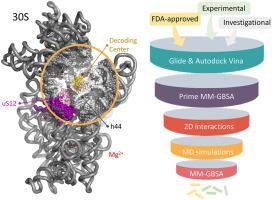
The global antibiotic resistance problem necessitates fast and effective approaches to finding novel inhibitors to treat bacterial infections. In this study, we propose a computational workflow to identify plausible high-affinity compounds from FDA-approved, investigational, and experimental libraries for the decoding center on the small subunit 30S of the E. coli ribosome. The workflow basically consists of two molecular docking calculations on the intact 30S, followed by molecular dynamics (MD) simulations coupled with MM-GBSA calculations on a truncated ribosome structure. The parameters used in the molecular docking suits, Glide and AutoDock Vina, as well as in the MD simulations with Desmond were carefully adjusted to obtain expected interactions for the ligand-rRNA complexes. A filtering procedure was followed, considering a fingerprint based on aminoglycoside's binding site on the 30S to obtain seven hit compounds either with different clinical usages or aminoglycoside derivatives under investigation, suggested for in vitro studies. The detailed workflow developed in this study promises an effective and fast approach for the estimation of binding free energies of large protein-RNA and ligand complexes.
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


