María Alcázar Fabra, Abraham J. Paredes-Fuentes, Manuel Torralba Carnerero, Daniel J. Moreno Férnandez de Ayala, Antonio Arroyo Luque, Ana Sánchez Cuesta, Carmine Staiano, Paula Sanchez-Pintos, María Luz Couce, Miguel Tomás, Ana Victoria Marco-Hernández, Carmen Orellana, Francisco Martínez, Mónica Roselló, Alfonso Caro, Juan Silvestre Oltra Soler, Sandra Monfort, Alejandro Sánchez, Dolores Rausell, Isidro Vitoria, Mireia del Toro, Angels Garcia-Cazorla, Natalia A. Julia-Palacios, Cristina Jou, Delia Yubero, Luis Carlos López, Juan Diego Hernández Camacho, Guillermo López Lluch, Manuel Ballesteros Simarro, Juan Carlos Rodríguez Aguilera, Gloria Brea Calvo, María Victoria Cascajo Almenara, Rafael Artuch, Carlos Santos-Ocaña
{"title":"新变体扩大了 COQ7 缺乏症的神经表型。","authors":"María Alcázar Fabra, Abraham J. Paredes-Fuentes, Manuel Torralba Carnerero, Daniel J. Moreno Férnandez de Ayala, Antonio Arroyo Luque, Ana Sánchez Cuesta, Carmine Staiano, Paula Sanchez-Pintos, María Luz Couce, Miguel Tomás, Ana Victoria Marco-Hernández, Carmen Orellana, Francisco Martínez, Mónica Roselló, Alfonso Caro, Juan Silvestre Oltra Soler, Sandra Monfort, Alejandro Sánchez, Dolores Rausell, Isidro Vitoria, Mireia del Toro, Angels Garcia-Cazorla, Natalia A. Julia-Palacios, Cristina Jou, Delia Yubero, Luis Carlos López, Juan Diego Hernández Camacho, Guillermo López Lluch, Manuel Ballesteros Simarro, Juan Carlos Rodríguez Aguilera, Gloria Brea Calvo, María Victoria Cascajo Almenara, Rafael Artuch, Carlos Santos-Ocaña","doi":"10.1002/jimd.12776","DOIUrl":null,"url":null,"abstract":"<p>The protein encoded by <i>COQ7</i> is required for CoQ<sub>10</sub> synthesis in humans, hydroxylating 3-demethoxyubiquinol (DMQ<sub>10</sub>) in the second to last steps of the pathway. <i>COQ7</i> mutations lead to a primary CoQ<sub>10</sub> deficiency syndrome associated with a pleiotropic neurological disorder. This study shows the clinical, physiological, and molecular characterization of four new cases of CoQ<sub>10</sub> primary deficiency caused by five mutations in <i>COQ7</i>, three of which have not yet been described, inducing mitochondrial dysfunction in all patients. However, the specific combination of the identified variants in each patient generated precise pathophysiological and molecular alterations in fibroblasts, which would explain the differential in vitro response to supplementation therapy. Our results suggest that COQ7 dysfunction could be caused by specific structural changes that affect the interaction with COQ9 required for the DMQ<sub>10</sub> presentation to COQ7, the substrate access to the active site, and the maintenance of the active site structure. Remarkably, patients' fibroblasts share transcriptional remodeling, supporting a modification of energy metabolism towards glycolysis, which could be an adaptive mechanism against CoQ<sub>10</sub> deficiency. However, transcriptional analysis of mitochondria-associated pathways showed distinct and dramatic differences between patient fibroblasts, which correlated with the extent of pathophysiological and neurological alterations observed in the probands. Overall, this study suggests that the combination of precise genetic diagnostics and the availability of new structural models of human proteins could help explain the origin of phenotypic pleiotropy observed in some genetic diseases and the different responses to available therapies.</p>","PeriodicalId":16281,"journal":{"name":"Journal of Inherited Metabolic Disease","volume":"47 5","pages":"1047-1068"},"PeriodicalIF":4.2000,"publicationDate":"2024-07-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jimd.12776","citationCount":"0","resultStr":"{\"title\":\"New variants expand the neurological phenotype of COQ7 deficiency\",\"authors\":\"María Alcázar Fabra, Abraham J. Paredes-Fuentes, Manuel Torralba Carnerero, Daniel J. Moreno Férnandez de Ayala, Antonio Arroyo Luque, Ana Sánchez Cuesta, Carmine Staiano, Paula Sanchez-Pintos, María Luz Couce, Miguel Tomás, Ana Victoria Marco-Hernández, Carmen Orellana, Francisco Martínez, Mónica Roselló, Alfonso Caro, Juan Silvestre Oltra Soler, Sandra Monfort, Alejandro Sánchez, Dolores Rausell, Isidro Vitoria, Mireia del Toro, Angels Garcia-Cazorla, Natalia A. Julia-Palacios, Cristina Jou, Delia Yubero, Luis Carlos López, Juan Diego Hernández Camacho, Guillermo López Lluch, Manuel Ballesteros Simarro, Juan Carlos Rodríguez Aguilera, Gloria Brea Calvo, María Victoria Cascajo Almenara, Rafael Artuch, Carlos Santos-Ocaña\",\"doi\":\"10.1002/jimd.12776\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The protein encoded by <i>COQ7</i> is required for CoQ<sub>10</sub> synthesis in humans, hydroxylating 3-demethoxyubiquinol (DMQ<sub>10</sub>) in the second to last steps of the pathway. <i>COQ7</i> mutations lead to a primary CoQ<sub>10</sub> deficiency syndrome associated with a pleiotropic neurological disorder. This study shows the clinical, physiological, and molecular characterization of four new cases of CoQ<sub>10</sub> primary deficiency caused by five mutations in <i>COQ7</i>, three of which have not yet been described, inducing mitochondrial dysfunction in all patients. However, the specific combination of the identified variants in each patient generated precise pathophysiological and molecular alterations in fibroblasts, which would explain the differential in vitro response to supplementation therapy. Our results suggest that COQ7 dysfunction could be caused by specific structural changes that affect the interaction with COQ9 required for the DMQ<sub>10</sub> presentation to COQ7, the substrate access to the active site, and the maintenance of the active site structure. Remarkably, patients' fibroblasts share transcriptional remodeling, supporting a modification of energy metabolism towards glycolysis, which could be an adaptive mechanism against CoQ<sub>10</sub> deficiency. However, transcriptional analysis of mitochondria-associated pathways showed distinct and dramatic differences between patient fibroblasts, which correlated with the extent of pathophysiological and neurological alterations observed in the probands. Overall, this study suggests that the combination of precise genetic diagnostics and the availability of new structural models of human proteins could help explain the origin of phenotypic pleiotropy observed in some genetic diseases and the different responses to available therapies.</p>\",\"PeriodicalId\":16281,\"journal\":{\"name\":\"Journal of Inherited Metabolic Disease\",\"volume\":\"47 5\",\"pages\":\"1047-1068\"},\"PeriodicalIF\":4.2000,\"publicationDate\":\"2024-07-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jimd.12776\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Inherited Metabolic Disease\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jimd.12776\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Inherited Metabolic Disease","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jimd.12776","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
New variants expand the neurological phenotype of COQ7 deficiency
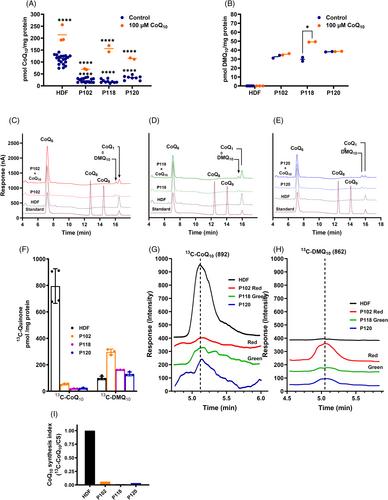
The protein encoded by COQ7 is required for CoQ10 synthesis in humans, hydroxylating 3-demethoxyubiquinol (DMQ10) in the second to last steps of the pathway. COQ7 mutations lead to a primary CoQ10 deficiency syndrome associated with a pleiotropic neurological disorder. This study shows the clinical, physiological, and molecular characterization of four new cases of CoQ10 primary deficiency caused by five mutations in COQ7, three of which have not yet been described, inducing mitochondrial dysfunction in all patients. However, the specific combination of the identified variants in each patient generated precise pathophysiological and molecular alterations in fibroblasts, which would explain the differential in vitro response to supplementation therapy. Our results suggest that COQ7 dysfunction could be caused by specific structural changes that affect the interaction with COQ9 required for the DMQ10 presentation to COQ7, the substrate access to the active site, and the maintenance of the active site structure. Remarkably, patients' fibroblasts share transcriptional remodeling, supporting a modification of energy metabolism towards glycolysis, which could be an adaptive mechanism against CoQ10 deficiency. However, transcriptional analysis of mitochondria-associated pathways showed distinct and dramatic differences between patient fibroblasts, which correlated with the extent of pathophysiological and neurological alterations observed in the probands. Overall, this study suggests that the combination of precise genetic diagnostics and the availability of new structural models of human proteins could help explain the origin of phenotypic pleiotropy observed in some genetic diseases and the different responses to available therapies.
期刊介绍:
The Journal of Inherited Metabolic Disease (JIMD) is the official journal of the Society for the Study of Inborn Errors of Metabolism (SSIEM). By enhancing communication between workers in the field throughout the world, the JIMD aims to improve the management and understanding of inherited metabolic disorders. It publishes results of original research and new or important observations pertaining to any aspect of inherited metabolic disease in humans and higher animals. This includes clinical (medical, dental and veterinary), biochemical, genetic (including cytogenetic, molecular and population genetic), experimental (including cell biological), methodological, theoretical, epidemiological, ethical and counselling aspects. The JIMD also reviews important new developments or controversial issues relating to metabolic disorders and publishes reviews and short reports arising from the Society''s annual symposia. A distinction is made between peer-reviewed scientific material that is selected because of its significance for other professionals in the field and non-peer- reviewed material that aims to be important, controversial, interesting or entertaining (“Extras”).

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


