Preeya Rehsi, Karolina Witek, Erin Emmett, Rachel Carling, Charles Turner, Neil Dalton, Tim Hutchin, Nedim Hadzic, Anil Dhawan, Roshni Vara
{"title":"缺乏琥珀酰丙酮的遗传性酪氨酸血症 1 型:4-氧代 6-羟基己酸酯 (4OHHA),一种潜在的诊断生物标志物","authors":"Preeya Rehsi, Karolina Witek, Erin Emmett, Rachel Carling, Charles Turner, Neil Dalton, Tim Hutchin, Nedim Hadzic, Anil Dhawan, Roshni Vara","doi":"10.1002/jmd2.12436","DOIUrl":null,"url":null,"abstract":"<p>Hereditary tyrosinemia type 1 (HT1) is a rare metabolic disease resulting in acute liver failure in early infancy, hypophosphataemic rickets, neurological crises, liver cirrhosis and risk of hepatocellular carcinoma later on in life. It is caused by the deficiency of the enzyme fumarylacetoacetate hydrolase which is involved in the terminal step of the catabolic pathway of tyrosine. Diagnosis is made through clinical suspicion supported by biochemical abnormalities that result from accumulation of upstream metabolites. Detection of succinylacetone (SA) in dried blood spot or urine remains pathognomonic, however it is not always detectable. Here we describe three cases of HT1 presenting with atypical biochemistry, where SA was not always detectable, highlighting the importance of an additional disease biomarker, 4-oxo-6-hydroxyheptanoate.</p>","PeriodicalId":14930,"journal":{"name":"JIMD reports","volume":"65 4","pages":"255-261"},"PeriodicalIF":1.8000,"publicationDate":"2024-06-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jmd2.12436","citationCount":"0","resultStr":"{\"title\":\"Hereditary tyrosinaemia type 1 in the absence of succinylacetone: 4-oxo 6-hydroxyhepanoate (4OHHA), a putative diagnostic biomarker\",\"authors\":\"Preeya Rehsi, Karolina Witek, Erin Emmett, Rachel Carling, Charles Turner, Neil Dalton, Tim Hutchin, Nedim Hadzic, Anil Dhawan, Roshni Vara\",\"doi\":\"10.1002/jmd2.12436\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Hereditary tyrosinemia type 1 (HT1) is a rare metabolic disease resulting in acute liver failure in early infancy, hypophosphataemic rickets, neurological crises, liver cirrhosis and risk of hepatocellular carcinoma later on in life. It is caused by the deficiency of the enzyme fumarylacetoacetate hydrolase which is involved in the terminal step of the catabolic pathway of tyrosine. Diagnosis is made through clinical suspicion supported by biochemical abnormalities that result from accumulation of upstream metabolites. Detection of succinylacetone (SA) in dried blood spot or urine remains pathognomonic, however it is not always detectable. Here we describe three cases of HT1 presenting with atypical biochemistry, where SA was not always detectable, highlighting the importance of an additional disease biomarker, 4-oxo-6-hydroxyheptanoate.</p>\",\"PeriodicalId\":14930,\"journal\":{\"name\":\"JIMD reports\",\"volume\":\"65 4\",\"pages\":\"255-261\"},\"PeriodicalIF\":1.8000,\"publicationDate\":\"2024-06-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jmd2.12436\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"JIMD reports\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jmd2.12436\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"JIMD reports","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jmd2.12436","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
Hereditary tyrosinaemia type 1 in the absence of succinylacetone: 4-oxo 6-hydroxyhepanoate (4OHHA), a putative diagnostic biomarker
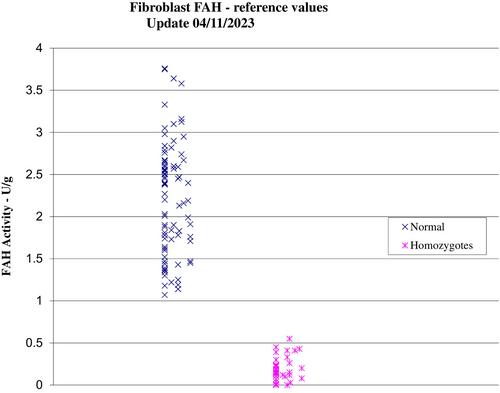
Hereditary tyrosinemia type 1 (HT1) is a rare metabolic disease resulting in acute liver failure in early infancy, hypophosphataemic rickets, neurological crises, liver cirrhosis and risk of hepatocellular carcinoma later on in life. It is caused by the deficiency of the enzyme fumarylacetoacetate hydrolase which is involved in the terminal step of the catabolic pathway of tyrosine. Diagnosis is made through clinical suspicion supported by biochemical abnormalities that result from accumulation of upstream metabolites. Detection of succinylacetone (SA) in dried blood spot or urine remains pathognomonic, however it is not always detectable. Here we describe three cases of HT1 presenting with atypical biochemistry, where SA was not always detectable, highlighting the importance of an additional disease biomarker, 4-oxo-6-hydroxyheptanoate.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


