{"title":"基于基因组变异和深度学习追溯地理种群的谱系起源。","authors":"Bing Yang , Xin Zhou , Shanlin Liu","doi":"10.1016/j.ympev.2024.108142","DOIUrl":null,"url":null,"abstract":"<div><p>Assigning a query individual animal or plant to its derived population is a prime task in diverse applications related to organismal genealogy. Such endeavors have conventionally relied on short DNA sequences under a phylogenetic framework. These methods naturally show constraints when the inferred population sources are ambiguously phylogenetically structured, a scenario demanding substantially more informative genetic signals. Recent advances in cost-effective production of whole-genome sequences and artificial intelligence have created an unprecedented opportunity to trace the population origin for essentially any given individual, as long as the genome reference data are comprehensive and standardized. Here, we developed a convolutional neural network method to identify population origins using genomic SNPs. Three empirical datasets (an Asian honeybee, a red fire ant, and a chicken datasets) and two simulated populations are used for the proof of concepts. The performance tests indicate that our method can accurately identify the genealogy origin of query individuals, with success rates ranging from 93 % to 100 %. We further showed that the accuracy of the model can be significantly increased by refining the informative sites through <span><math><mrow><msub><mi>F</mi><mrow><mi>ST</mi></mrow></msub></mrow></math></span> filtering. Our method is robust to configurations related to batch sizes and epochs, whereas model learning benefits from the setting of a proper preset learning rate. Moreover, we explained the importance score of key sites for algorithm interpretability and credibility, which has been largely ignored. We anticipate that by coupling genomics and deep learning, our method will see broad potential in conservation and management applications that involve natural resources, invasive pests and weeds, and illegal trades of wildlife products.</p></div>","PeriodicalId":56109,"journal":{"name":"Molecular Phylogenetics and Evolution","volume":"198 ","pages":"Article 108142"},"PeriodicalIF":3.6000,"publicationDate":"2024-07-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Tracing the genealogy origin of geographic populations based on genomic variation and deep learning\",\"authors\":\"Bing Yang , Xin Zhou , Shanlin Liu\",\"doi\":\"10.1016/j.ympev.2024.108142\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Assigning a query individual animal or plant to its derived population is a prime task in diverse applications related to organismal genealogy. Such endeavors have conventionally relied on short DNA sequences under a phylogenetic framework. These methods naturally show constraints when the inferred population sources are ambiguously phylogenetically structured, a scenario demanding substantially more informative genetic signals. Recent advances in cost-effective production of whole-genome sequences and artificial intelligence have created an unprecedented opportunity to trace the population origin for essentially any given individual, as long as the genome reference data are comprehensive and standardized. Here, we developed a convolutional neural network method to identify population origins using genomic SNPs. Three empirical datasets (an Asian honeybee, a red fire ant, and a chicken datasets) and two simulated populations are used for the proof of concepts. The performance tests indicate that our method can accurately identify the genealogy origin of query individuals, with success rates ranging from 93 % to 100 %. We further showed that the accuracy of the model can be significantly increased by refining the informative sites through <span><math><mrow><msub><mi>F</mi><mrow><mi>ST</mi></mrow></msub></mrow></math></span> filtering. Our method is robust to configurations related to batch sizes and epochs, whereas model learning benefits from the setting of a proper preset learning rate. Moreover, we explained the importance score of key sites for algorithm interpretability and credibility, which has been largely ignored. We anticipate that by coupling genomics and deep learning, our method will see broad potential in conservation and management applications that involve natural resources, invasive pests and weeds, and illegal trades of wildlife products.</p></div>\",\"PeriodicalId\":56109,\"journal\":{\"name\":\"Molecular Phylogenetics and Evolution\",\"volume\":\"198 \",\"pages\":\"Article 108142\"},\"PeriodicalIF\":3.6000,\"publicationDate\":\"2024-07-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Phylogenetics and Evolution\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S1055790324001349\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Phylogenetics and Evolution","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1055790324001349","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Tracing the genealogy origin of geographic populations based on genomic variation and deep learning
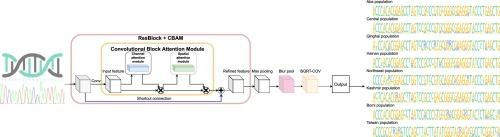
Assigning a query individual animal or plant to its derived population is a prime task in diverse applications related to organismal genealogy. Such endeavors have conventionally relied on short DNA sequences under a phylogenetic framework. These methods naturally show constraints when the inferred population sources are ambiguously phylogenetically structured, a scenario demanding substantially more informative genetic signals. Recent advances in cost-effective production of whole-genome sequences and artificial intelligence have created an unprecedented opportunity to trace the population origin for essentially any given individual, as long as the genome reference data are comprehensive and standardized. Here, we developed a convolutional neural network method to identify population origins using genomic SNPs. Three empirical datasets (an Asian honeybee, a red fire ant, and a chicken datasets) and two simulated populations are used for the proof of concepts. The performance tests indicate that our method can accurately identify the genealogy origin of query individuals, with success rates ranging from 93 % to 100 %. We further showed that the accuracy of the model can be significantly increased by refining the informative sites through filtering. Our method is robust to configurations related to batch sizes and epochs, whereas model learning benefits from the setting of a proper preset learning rate. Moreover, we explained the importance score of key sites for algorithm interpretability and credibility, which has been largely ignored. We anticipate that by coupling genomics and deep learning, our method will see broad potential in conservation and management applications that involve natural resources, invasive pests and weeds, and illegal trades of wildlife products.
期刊介绍:
Molecular Phylogenetics and Evolution is dedicated to bringing Darwin''s dream within grasp - to "have fairly true genealogical trees of each great kingdom of Nature." The journal provides a forum for molecular studies that advance our understanding of phylogeny and evolution, further the development of phylogenetically more accurate taxonomic classifications, and ultimately bring a unified classification for all the ramifying lines of life. Phylogeographic studies will be considered for publication if they offer EXCEPTIONAL theoretical or empirical advances.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


