N. A. Kryuchkova, A. I. Stadnichenko, E. V. Korotaev, V. V. Krisyuk
{"title":"以异性铜(II)配合物为例,研究 MOCVD 前驱体的电子结构和分子重排模拟,以预测其蒸发时的热稳定性","authors":"N. A. Kryuchkova, A. I. Stadnichenko, E. V. Korotaev, V. V. Krisyuk","doi":"10.1134/S0022476624050044","DOIUrl":null,"url":null,"abstract":"<p>An approach for predicting stability of organometallic precursors during evaporation for chemical vapor deposition is considered on the example of isostructural heteroleptic copper complexes [Cu(acac)(hfac)]<sub>2</sub> (<b>1</b>) and [Cu(ki)(hfac)]<sub>2</sub> (<b>2</b>). The electron density distribution in binuclear molecules of <b>1</b> and <b>2</b> is studied by the density functional theory (DFT) and X-ray photoelectron spectroscopy (XPS). It is established that the highest occupied molecular orbitals (HOMOs) and the lowest unoccupied molecular orbitals (LUMOs) of these complexes are characterized by the same composition and structure, their metal–ligand bonds and bridging Cu–O bonds in dimers have close energies, and their donor and central atoms have equal charges. It is shown that the differences between resistances of the studied heteroleptic complexes to the disproportionation upon condensed-phase heating, leading to the formation of homoleptic complexes, are determined by the kinetics of the process. We propose a mechanism of thermally activated ligand-exchange reaction as a series of rearrangements of dimeric complexes in crystals. It is shown from the calculated Δ<i>E</i> and Δ<i>G</i> values that <b>2</b> is thermally more stable than <b>1</b> due to the presence of an energy barriers it encounters at each stage of the process.</p>","PeriodicalId":668,"journal":{"name":"Journal of Structural Chemistry","volume":"65 5","pages":"895 - 906"},"PeriodicalIF":1.4000,"publicationDate":"2024-06-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Study of Electronic Structure and Simulation of Molecular Rearrangements of MOCVD Precursors to Predict Their Thermal Stability Upon Evaporation on the Example of Heteroleptic Copper(II) Complexes\",\"authors\":\"N. A. Kryuchkova, A. I. Stadnichenko, E. V. Korotaev, V. V. Krisyuk\",\"doi\":\"10.1134/S0022476624050044\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>An approach for predicting stability of organometallic precursors during evaporation for chemical vapor deposition is considered on the example of isostructural heteroleptic copper complexes [Cu(acac)(hfac)]<sub>2</sub> (<b>1</b>) and [Cu(ki)(hfac)]<sub>2</sub> (<b>2</b>). The electron density distribution in binuclear molecules of <b>1</b> and <b>2</b> is studied by the density functional theory (DFT) and X-ray photoelectron spectroscopy (XPS). It is established that the highest occupied molecular orbitals (HOMOs) and the lowest unoccupied molecular orbitals (LUMOs) of these complexes are characterized by the same composition and structure, their metal–ligand bonds and bridging Cu–O bonds in dimers have close energies, and their donor and central atoms have equal charges. It is shown that the differences between resistances of the studied heteroleptic complexes to the disproportionation upon condensed-phase heating, leading to the formation of homoleptic complexes, are determined by the kinetics of the process. We propose a mechanism of thermally activated ligand-exchange reaction as a series of rearrangements of dimeric complexes in crystals. It is shown from the calculated Δ<i>E</i> and Δ<i>G</i> values that <b>2</b> is thermally more stable than <b>1</b> due to the presence of an energy barriers it encounters at each stage of the process.</p>\",\"PeriodicalId\":668,\"journal\":{\"name\":\"Journal of Structural Chemistry\",\"volume\":\"65 5\",\"pages\":\"895 - 906\"},\"PeriodicalIF\":1.4000,\"publicationDate\":\"2024-06-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Structural Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://link.springer.com/article/10.1134/S0022476624050044\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"CHEMISTRY, INORGANIC & NUCLEAR\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Structural Chemistry","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1134/S0022476624050044","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"CHEMISTRY, INORGANIC & NUCLEAR","Score":null,"Total":0}
Study of Electronic Structure and Simulation of Molecular Rearrangements of MOCVD Precursors to Predict Their Thermal Stability Upon Evaporation on the Example of Heteroleptic Copper(II) Complexes
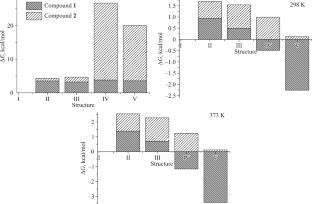
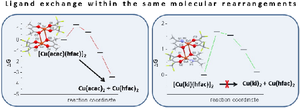
An approach for predicting stability of organometallic precursors during evaporation for chemical vapor deposition is considered on the example of isostructural heteroleptic copper complexes [Cu(acac)(hfac)]2 (1) and [Cu(ki)(hfac)]2 (2). The electron density distribution in binuclear molecules of 1 and 2 is studied by the density functional theory (DFT) and X-ray photoelectron spectroscopy (XPS). It is established that the highest occupied molecular orbitals (HOMOs) and the lowest unoccupied molecular orbitals (LUMOs) of these complexes are characterized by the same composition and structure, their metal–ligand bonds and bridging Cu–O bonds in dimers have close energies, and their donor and central atoms have equal charges. It is shown that the differences between resistances of the studied heteroleptic complexes to the disproportionation upon condensed-phase heating, leading to the formation of homoleptic complexes, are determined by the kinetics of the process. We propose a mechanism of thermally activated ligand-exchange reaction as a series of rearrangements of dimeric complexes in crystals. It is shown from the calculated ΔE and ΔG values that 2 is thermally more stable than 1 due to the presence of an energy barriers it encounters at each stage of the process.
期刊介绍:
Journal is an interdisciplinary publication covering all aspects of structural chemistry, including the theory of molecular structure and chemical bond; the use of physical methods to study the electronic and spatial structure of chemical species; structural features of liquids, solutions, surfaces, supramolecular systems, nano- and solid materials; and the crystal structure of solids.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


