{"title":"mACPpred 2.0:利用集成空间和概率特征表征的堆叠深度学习进行抗癌肽预测","authors":"","doi":"10.1016/j.jmb.2024.168687","DOIUrl":null,"url":null,"abstract":"<div><p>Anticancer peptides (ACPs), naturally occurring molecules with remarkable potential to target and kill cancer cells. However, identifying ACPs based solely from their primary amino acid sequences remains a major hurdle in immunoinformatics. In the past, several web-based machine learning (ML) tools have been proposed to assist researchers in identifying potential ACPs for further testing. Notably, our meta-approach method, mACPpred, introduced in 2019, has significantly advanced the field of ACP research. Given the exponential growth in the number of characterized ACPs, there is now a pressing need to create an updated version of mACPpred. To develop mACPpred 2.0, we constructed an up-to-date benchmarking dataset by integrating all publicly available ACP datasets. We employed a large-scale of feature descriptors, encompassing both conventional feature descriptors and advanced pre-trained natural language processing (NLP)-based embeddings. We evaluated their ability to discriminate between ACPs and non-ACPs using eleven different classifiers. Subsequently, we employed a stacked deep learning (SDL) approach, incorporating 1D convolutional neural network (1D CNN) blocks and hybrid features. These features included the top seven performing NLP-based features and 90 probabilistic features, allowing us to identify hidden patterns within these diverse features and improve the accuracy of our ACP prediction model. This is the first study to integrate spatial and probabilistic feature representations for predicting ACPs. Rigorous cross-validation and independent tests conclusively demonstrated that mACPpred 2.0 not only surpassed its predecessor (mACPpred) but also outperformed the existing state-of-the-art predictors, highlighting the importance of advanced feature representation capabilities attained through SDL. To facilitate widespread use and accessibility, we have developed a user-friendly for mACPpred 2.0, available at <span><span>https://balalab-skku.org/mACPpred2/</span><svg><path></path></svg></span>.</p></div>","PeriodicalId":369,"journal":{"name":"Journal of Molecular Biology","volume":"436 17","pages":"Article 168687"},"PeriodicalIF":4.7000,"publicationDate":"2024-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S0022283624002894/pdfft?md5=ecdf80bb684910ec5433145962a8f247&pid=1-s2.0-S0022283624002894-main.pdf","citationCount":"0","resultStr":"{\"title\":\"mACPpred 2.0: Stacked Deep Learning for Anticancer Peptide Prediction with Integrated Spatial and Probabilistic Feature Representations\",\"authors\":\"\",\"doi\":\"10.1016/j.jmb.2024.168687\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Anticancer peptides (ACPs), naturally occurring molecules with remarkable potential to target and kill cancer cells. However, identifying ACPs based solely from their primary amino acid sequences remains a major hurdle in immunoinformatics. In the past, several web-based machine learning (ML) tools have been proposed to assist researchers in identifying potential ACPs for further testing. Notably, our meta-approach method, mACPpred, introduced in 2019, has significantly advanced the field of ACP research. Given the exponential growth in the number of characterized ACPs, there is now a pressing need to create an updated version of mACPpred. To develop mACPpred 2.0, we constructed an up-to-date benchmarking dataset by integrating all publicly available ACP datasets. We employed a large-scale of feature descriptors, encompassing both conventional feature descriptors and advanced pre-trained natural language processing (NLP)-based embeddings. We evaluated their ability to discriminate between ACPs and non-ACPs using eleven different classifiers. Subsequently, we employed a stacked deep learning (SDL) approach, incorporating 1D convolutional neural network (1D CNN) blocks and hybrid features. These features included the top seven performing NLP-based features and 90 probabilistic features, allowing us to identify hidden patterns within these diverse features and improve the accuracy of our ACP prediction model. This is the first study to integrate spatial and probabilistic feature representations for predicting ACPs. Rigorous cross-validation and independent tests conclusively demonstrated that mACPpred 2.0 not only surpassed its predecessor (mACPpred) but also outperformed the existing state-of-the-art predictors, highlighting the importance of advanced feature representation capabilities attained through SDL. To facilitate widespread use and accessibility, we have developed a user-friendly for mACPpred 2.0, available at <span><span>https://balalab-skku.org/mACPpred2/</span><svg><path></path></svg></span>.</p></div>\",\"PeriodicalId\":369,\"journal\":{\"name\":\"Journal of Molecular Biology\",\"volume\":\"436 17\",\"pages\":\"Article 168687\"},\"PeriodicalIF\":4.7000,\"publicationDate\":\"2024-09-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.sciencedirect.com/science/article/pii/S0022283624002894/pdfft?md5=ecdf80bb684910ec5433145962a8f247&pid=1-s2.0-S0022283624002894-main.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Molecular Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0022283624002894\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Biology","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0022283624002894","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
mACPpred 2.0: Stacked Deep Learning for Anticancer Peptide Prediction with Integrated Spatial and Probabilistic Feature Representations
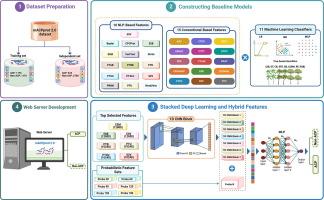
Anticancer peptides (ACPs), naturally occurring molecules with remarkable potential to target and kill cancer cells. However, identifying ACPs based solely from their primary amino acid sequences remains a major hurdle in immunoinformatics. In the past, several web-based machine learning (ML) tools have been proposed to assist researchers in identifying potential ACPs for further testing. Notably, our meta-approach method, mACPpred, introduced in 2019, has significantly advanced the field of ACP research. Given the exponential growth in the number of characterized ACPs, there is now a pressing need to create an updated version of mACPpred. To develop mACPpred 2.0, we constructed an up-to-date benchmarking dataset by integrating all publicly available ACP datasets. We employed a large-scale of feature descriptors, encompassing both conventional feature descriptors and advanced pre-trained natural language processing (NLP)-based embeddings. We evaluated their ability to discriminate between ACPs and non-ACPs using eleven different classifiers. Subsequently, we employed a stacked deep learning (SDL) approach, incorporating 1D convolutional neural network (1D CNN) blocks and hybrid features. These features included the top seven performing NLP-based features and 90 probabilistic features, allowing us to identify hidden patterns within these diverse features and improve the accuracy of our ACP prediction model. This is the first study to integrate spatial and probabilistic feature representations for predicting ACPs. Rigorous cross-validation and independent tests conclusively demonstrated that mACPpred 2.0 not only surpassed its predecessor (mACPpred) but also outperformed the existing state-of-the-art predictors, highlighting the importance of advanced feature representation capabilities attained through SDL. To facilitate widespread use and accessibility, we have developed a user-friendly for mACPpred 2.0, available at https://balalab-skku.org/mACPpred2/.
期刊介绍:
Journal of Molecular Biology (JMB) provides high quality, comprehensive and broad coverage in all areas of molecular biology. The journal publishes original scientific research papers that provide mechanistic and functional insights and report a significant advance to the field. The journal encourages the submission of multidisciplinary studies that use complementary experimental and computational approaches to address challenging biological questions.
Research areas include but are not limited to: Biomolecular interactions, signaling networks, systems biology; Cell cycle, cell growth, cell differentiation; Cell death, autophagy; Cell signaling and regulation; Chemical biology; Computational biology, in combination with experimental studies; DNA replication, repair, and recombination; Development, regenerative biology, mechanistic and functional studies of stem cells; Epigenetics, chromatin structure and function; Gene expression; Membrane processes, cell surface proteins and cell-cell interactions; Methodological advances, both experimental and theoretical, including databases; Microbiology, virology, and interactions with the host or environment; Microbiota mechanistic and functional studies; Nuclear organization; Post-translational modifications, proteomics; Processing and function of biologically important macromolecules and complexes; Molecular basis of disease; RNA processing, structure and functions of non-coding RNAs, transcription; Sorting, spatiotemporal organization, trafficking; Structural biology; Synthetic biology; Translation, protein folding, chaperones, protein degradation and quality control.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


