Emma Rydholm, Tomas Bastys, Emma Svensson, Christos Kannas, Ola Engkvist and Thierry Kogej
{"title":"利用化学反应知识图谱拓展化学空间","authors":"Emma Rydholm, Tomas Bastys, Emma Svensson, Christos Kannas, Ola Engkvist and Thierry Kogej","doi":"10.1039/D3DD00230F","DOIUrl":null,"url":null,"abstract":"<p >In this work, we present a new molecular <em>de novo</em> design approach which utilizes a knowledge graph encoding chemical reactions, extracted from the publicly available USPTO (United States Patent and Trademark Office) dataset. Our proposed method can be used to expand the chemical space by performing forward synthesis prediction by finding new combinations of reactants in the knowledge graph and can in this way generate libraries of <em>de novo</em> compounds along with a valid synthetic route. The forward synthesis prediction of novel compounds involves two steps. In the first step, a graph neural network-based link prediction model is used to suggest pairs of existing reactant nodes in the graph that are likely to react. In the second step, product prediction is performed using a molecular transformer model to obtain the potential products for the suggested reactant pairs. We achieve a ROC–AUC score of 0.861 for link prediction in the knowledge graph and for the product prediction, a top-1 accuracy of 0.924. The method's utility is demonstrated by generating a set of <em>de novo</em> compounds by predicting high probability reactions in the USPTO. The generated compounds are diverse in nature and many exhibit drug-like properties. A brief comparison with a template-based library design is provided. Furthermore, evaluation of the potential activity using a quantitative structure–activity relationship (QSAR) model suggested the presence of potential dopamine receptor D2 (DRD2) modulators among the proposed compounds. In summary, our results suggest that the proposed method can expand the easily accessible chemical space, by combining known compounds, and identify novel drug-like compounds for a specific target.</p>","PeriodicalId":72816,"journal":{"name":"Digital discovery","volume":" 7","pages":" 1378-1388"},"PeriodicalIF":6.2000,"publicationDate":"2024-06-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2024/dd/d3dd00230f?page=search","citationCount":"0","resultStr":"{\"title\":\"Expanding the chemical space using a chemical reaction knowledge graph†\",\"authors\":\"Emma Rydholm, Tomas Bastys, Emma Svensson, Christos Kannas, Ola Engkvist and Thierry Kogej\",\"doi\":\"10.1039/D3DD00230F\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >In this work, we present a new molecular <em>de novo</em> design approach which utilizes a knowledge graph encoding chemical reactions, extracted from the publicly available USPTO (United States Patent and Trademark Office) dataset. Our proposed method can be used to expand the chemical space by performing forward synthesis prediction by finding new combinations of reactants in the knowledge graph and can in this way generate libraries of <em>de novo</em> compounds along with a valid synthetic route. The forward synthesis prediction of novel compounds involves two steps. In the first step, a graph neural network-based link prediction model is used to suggest pairs of existing reactant nodes in the graph that are likely to react. In the second step, product prediction is performed using a molecular transformer model to obtain the potential products for the suggested reactant pairs. We achieve a ROC–AUC score of 0.861 for link prediction in the knowledge graph and for the product prediction, a top-1 accuracy of 0.924. The method's utility is demonstrated by generating a set of <em>de novo</em> compounds by predicting high probability reactions in the USPTO. The generated compounds are diverse in nature and many exhibit drug-like properties. A brief comparison with a template-based library design is provided. Furthermore, evaluation of the potential activity using a quantitative structure–activity relationship (QSAR) model suggested the presence of potential dopamine receptor D2 (DRD2) modulators among the proposed compounds. In summary, our results suggest that the proposed method can expand the easily accessible chemical space, by combining known compounds, and identify novel drug-like compounds for a specific target.</p>\",\"PeriodicalId\":72816,\"journal\":{\"name\":\"Digital discovery\",\"volume\":\" 7\",\"pages\":\" 1378-1388\"},\"PeriodicalIF\":6.2000,\"publicationDate\":\"2024-06-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.rsc.org/en/content/articlepdf/2024/dd/d3dd00230f?page=search\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Digital discovery\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2024/dd/d3dd00230f\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Digital discovery","FirstCategoryId":"1085","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/dd/d3dd00230f","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Expanding the chemical space using a chemical reaction knowledge graph†
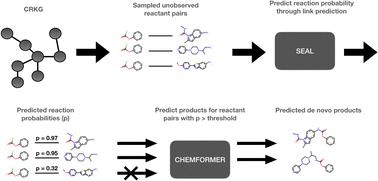
In this work, we present a new molecular de novo design approach which utilizes a knowledge graph encoding chemical reactions, extracted from the publicly available USPTO (United States Patent and Trademark Office) dataset. Our proposed method can be used to expand the chemical space by performing forward synthesis prediction by finding new combinations of reactants in the knowledge graph and can in this way generate libraries of de novo compounds along with a valid synthetic route. The forward synthesis prediction of novel compounds involves two steps. In the first step, a graph neural network-based link prediction model is used to suggest pairs of existing reactant nodes in the graph that are likely to react. In the second step, product prediction is performed using a molecular transformer model to obtain the potential products for the suggested reactant pairs. We achieve a ROC–AUC score of 0.861 for link prediction in the knowledge graph and for the product prediction, a top-1 accuracy of 0.924. The method's utility is demonstrated by generating a set of de novo compounds by predicting high probability reactions in the USPTO. The generated compounds are diverse in nature and many exhibit drug-like properties. A brief comparison with a template-based library design is provided. Furthermore, evaluation of the potential activity using a quantitative structure–activity relationship (QSAR) model suggested the presence of potential dopamine receptor D2 (DRD2) modulators among the proposed compounds. In summary, our results suggest that the proposed method can expand the easily accessible chemical space, by combining known compounds, and identify novel drug-like compounds for a specific target.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


