Matthew J. Kania, Albert Reyes and Sharon R. Neufeldt*,
{"title":"钯(0)上(异)芳基(假)卤化物的氧化加成:不同机理的起源和意义。","authors":"Matthew J. Kania, Albert Reyes and Sharon R. Neufeldt*, ","doi":"10.1021/jacs.4c04496","DOIUrl":null,"url":null,"abstract":"<p >Two limiting mechanisms are possible for oxidative addition of (hetero)aryl (pseudo)halides at Pd(0): a 3-centered concerted and a nucleophilic displacement mechanism. Until now, there has been little understanding about when each mechanism is relevant. Prior investigations to distinguish between these pathways were limited to a few specific combinations of the substrate and ligand. Here, we computationally evaluated over 180 transition structures for oxidative addition in order to determine mechanistic trends based on substrate, ligand(s), and coordination number. Natural abundance <sup>13</sup>C kinetic isotope effects provide experimental results consistent with computational predictions. Key findings include that (1) differences in highest occupied molecular orbital (HOMO) symmetries dictate that, although 12e<sup>–</sup> PdL is strongly biased toward a 3-centered concerted mechanism, 14e<sup>–</sup> PdL<sub>2</sub> often prefers a nucleophilic displacement mechanism; (2) ligand electronics and sterics, including ligand bite angle, influence the preferred mechanism of the reaction at PdL<sub>2</sub>; (3) phenyl triflate always reacts through a displacement mechanism regardless of the catalyst structure due to the stability of a triflate anion and the inability of oxygen to effectively donate electron density to Pd; and (4) the high reactivity of C–X bonds adjacent to nitrogen in pyridine substrates relates to stereoelectronic stabilization of a nucleophilic displacement transition state. This work has implications for controlling rate and selectivity in catalytic couplings, and we demonstrate application of the mechanistic insight toward chemodivergent cross-couplings of bromochloroheteroarenes.</p>","PeriodicalId":49,"journal":{"name":"Journal of the American Chemical Society","volume":null,"pages":null},"PeriodicalIF":14.4000,"publicationDate":"2024-07-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Oxidative Addition of (Hetero)aryl (Pseudo)halides at Palladium(0): Origin and Significance of Divergent Mechanisms\",\"authors\":\"Matthew J. Kania, Albert Reyes and Sharon R. Neufeldt*, \",\"doi\":\"10.1021/jacs.4c04496\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Two limiting mechanisms are possible for oxidative addition of (hetero)aryl (pseudo)halides at Pd(0): a 3-centered concerted and a nucleophilic displacement mechanism. Until now, there has been little understanding about when each mechanism is relevant. Prior investigations to distinguish between these pathways were limited to a few specific combinations of the substrate and ligand. Here, we computationally evaluated over 180 transition structures for oxidative addition in order to determine mechanistic trends based on substrate, ligand(s), and coordination number. Natural abundance <sup>13</sup>C kinetic isotope effects provide experimental results consistent with computational predictions. Key findings include that (1) differences in highest occupied molecular orbital (HOMO) symmetries dictate that, although 12e<sup>–</sup> PdL is strongly biased toward a 3-centered concerted mechanism, 14e<sup>–</sup> PdL<sub>2</sub> often prefers a nucleophilic displacement mechanism; (2) ligand electronics and sterics, including ligand bite angle, influence the preferred mechanism of the reaction at PdL<sub>2</sub>; (3) phenyl triflate always reacts through a displacement mechanism regardless of the catalyst structure due to the stability of a triflate anion and the inability of oxygen to effectively donate electron density to Pd; and (4) the high reactivity of C–X bonds adjacent to nitrogen in pyridine substrates relates to stereoelectronic stabilization of a nucleophilic displacement transition state. This work has implications for controlling rate and selectivity in catalytic couplings, and we demonstrate application of the mechanistic insight toward chemodivergent cross-couplings of bromochloroheteroarenes.</p>\",\"PeriodicalId\":49,\"journal\":{\"name\":\"Journal of the American Chemical Society\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":14.4000,\"publicationDate\":\"2024-07-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of the American Chemical Society\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/jacs.4c04496\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of the American Chemical Society","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/jacs.4c04496","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Oxidative Addition of (Hetero)aryl (Pseudo)halides at Palladium(0): Origin and Significance of Divergent Mechanisms
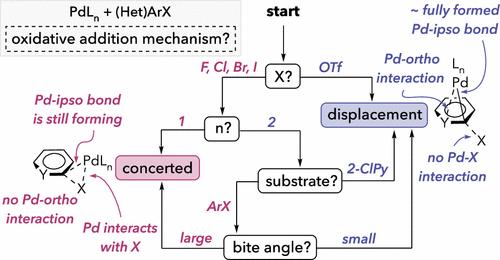
Two limiting mechanisms are possible for oxidative addition of (hetero)aryl (pseudo)halides at Pd(0): a 3-centered concerted and a nucleophilic displacement mechanism. Until now, there has been little understanding about when each mechanism is relevant. Prior investigations to distinguish between these pathways were limited to a few specific combinations of the substrate and ligand. Here, we computationally evaluated over 180 transition structures for oxidative addition in order to determine mechanistic trends based on substrate, ligand(s), and coordination number. Natural abundance 13C kinetic isotope effects provide experimental results consistent with computational predictions. Key findings include that (1) differences in highest occupied molecular orbital (HOMO) symmetries dictate that, although 12e– PdL is strongly biased toward a 3-centered concerted mechanism, 14e– PdL2 often prefers a nucleophilic displacement mechanism; (2) ligand electronics and sterics, including ligand bite angle, influence the preferred mechanism of the reaction at PdL2; (3) phenyl triflate always reacts through a displacement mechanism regardless of the catalyst structure due to the stability of a triflate anion and the inability of oxygen to effectively donate electron density to Pd; and (4) the high reactivity of C–X bonds adjacent to nitrogen in pyridine substrates relates to stereoelectronic stabilization of a nucleophilic displacement transition state. This work has implications for controlling rate and selectivity in catalytic couplings, and we demonstrate application of the mechanistic insight toward chemodivergent cross-couplings of bromochloroheteroarenes.
期刊介绍:
The flagship journal of the American Chemical Society, known as the Journal of the American Chemical Society (JACS), has been a prestigious publication since its establishment in 1879. It holds a preeminent position in the field of chemistry and related interdisciplinary sciences. JACS is committed to disseminating cutting-edge research papers, covering a wide range of topics, and encompasses approximately 19,000 pages of Articles, Communications, and Perspectives annually. With a weekly publication frequency, JACS plays a vital role in advancing the field of chemistry by providing essential research.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


