Darja Cvetković, Marija Mitrović Dankulov, Aleksandar Bogojević, Saša Lazović, Darija Obradović
{"title":"用分子方法和基于图表的方法加强汉森溶解度预测","authors":"Darja Cvetković, Marija Mitrović Dankulov, Aleksandar Bogojević, Saša Lazović, Darija Obradović","doi":"10.1016/j.chemolab.2024.105168","DOIUrl":null,"url":null,"abstract":"<div><p>The fast and accurate prediction of Hansen solubility benefits many diverse fields such as pharmaceuticals, the food industry, and cosmetics. To estimate the individual HSP values (polar, dispersive, and hydrogen bonding components), we investigated the performance of using Mordred descriptors in multiple linear regressions and XGBoost modeling. For HSP predictions, we also tested a graph-based molecular representation with graph neural network (GNN) modeling. To select the optimal models for final training and predictions, we used nested cross-validation and hyper-parameter optimization. The models with the best predictive performance were selected through internal (<em>R</em><sup><em>2</em></sup><sub>train</sub>, RMSE, MEPcv) and external (RMSEP, CCC, MEP, <em>R</em><sup><em>2</em></sup><sub>test</sub>, <em>ar</em><sup>2</sup>m, Δ<em>r</em><sup>2</sup>m) validation metrics using ∼1200 compounds from free-available database <span>https://www.stevenabbott.co.uk</span><svg><path></path></svg>. To confirm the practical reliability, we examined the agreement of experimentally obtained HSP data from the literature for 93 compounds and the data predicted by the created models. The results of GNN modeling showed the best predictive characteristics, which include a coefficient of determination between experimentally obtained and predicted HSP values greater than 0.76 for polar and hydrogen bond forces and greater than 0.66 for dispersive forces. Interpreting the fundamental basis of Hansen solubility using the created MLR equations and XGBoost models, HSP values were found to be influenced by van der Waals volume characteristics, 2D matrix molecular representation, and polarity. We elaborated on the practical benefits of using the selected GNN method through Hansen's solubility sphere as an example. This is the first study to demonstrate the advantages of GNN in predicting individual HSP components, as well as the first study to describe in detail their molecular basis using MLR and XGBoost modeling.</p></div>","PeriodicalId":9774,"journal":{"name":"Chemometrics and Intelligent Laboratory Systems","volume":"251 ","pages":"Article 105168"},"PeriodicalIF":3.7000,"publicationDate":"2024-06-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Enhancing Hansen Solubility Predictions with Molecular and Graph-Based Approaches\",\"authors\":\"Darja Cvetković, Marija Mitrović Dankulov, Aleksandar Bogojević, Saša Lazović, Darija Obradović\",\"doi\":\"10.1016/j.chemolab.2024.105168\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>The fast and accurate prediction of Hansen solubility benefits many diverse fields such as pharmaceuticals, the food industry, and cosmetics. To estimate the individual HSP values (polar, dispersive, and hydrogen bonding components), we investigated the performance of using Mordred descriptors in multiple linear regressions and XGBoost modeling. For HSP predictions, we also tested a graph-based molecular representation with graph neural network (GNN) modeling. To select the optimal models for final training and predictions, we used nested cross-validation and hyper-parameter optimization. The models with the best predictive performance were selected through internal (<em>R</em><sup><em>2</em></sup><sub>train</sub>, RMSE, MEPcv) and external (RMSEP, CCC, MEP, <em>R</em><sup><em>2</em></sup><sub>test</sub>, <em>ar</em><sup>2</sup>m, Δ<em>r</em><sup>2</sup>m) validation metrics using ∼1200 compounds from free-available database <span>https://www.stevenabbott.co.uk</span><svg><path></path></svg>. To confirm the practical reliability, we examined the agreement of experimentally obtained HSP data from the literature for 93 compounds and the data predicted by the created models. The results of GNN modeling showed the best predictive characteristics, which include a coefficient of determination between experimentally obtained and predicted HSP values greater than 0.76 for polar and hydrogen bond forces and greater than 0.66 for dispersive forces. Interpreting the fundamental basis of Hansen solubility using the created MLR equations and XGBoost models, HSP values were found to be influenced by van der Waals volume characteristics, 2D matrix molecular representation, and polarity. We elaborated on the practical benefits of using the selected GNN method through Hansen's solubility sphere as an example. This is the first study to demonstrate the advantages of GNN in predicting individual HSP components, as well as the first study to describe in detail their molecular basis using MLR and XGBoost modeling.</p></div>\",\"PeriodicalId\":9774,\"journal\":{\"name\":\"Chemometrics and Intelligent Laboratory Systems\",\"volume\":\"251 \",\"pages\":\"Article 105168\"},\"PeriodicalIF\":3.7000,\"publicationDate\":\"2024-06-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Chemometrics and Intelligent Laboratory Systems\",\"FirstCategoryId\":\"94\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0169743924001084\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"AUTOMATION & CONTROL SYSTEMS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Chemometrics and Intelligent Laboratory Systems","FirstCategoryId":"94","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0169743924001084","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"AUTOMATION & CONTROL SYSTEMS","Score":null,"Total":0}
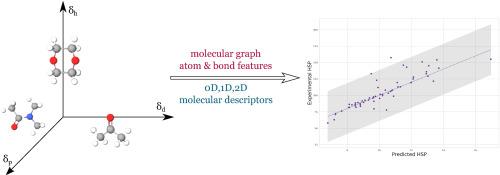
Enhancing Hansen Solubility Predictions with Molecular and Graph-Based Approaches
The fast and accurate prediction of Hansen solubility benefits many diverse fields such as pharmaceuticals, the food industry, and cosmetics. To estimate the individual HSP values (polar, dispersive, and hydrogen bonding components), we investigated the performance of using Mordred descriptors in multiple linear regressions and XGBoost modeling. For HSP predictions, we also tested a graph-based molecular representation with graph neural network (GNN) modeling. To select the optimal models for final training and predictions, we used nested cross-validation and hyper-parameter optimization. The models with the best predictive performance were selected through internal (R2train, RMSE, MEPcv) and external (RMSEP, CCC, MEP, R2test, ar2m, Δr2m) validation metrics using ∼1200 compounds from free-available database https://www.stevenabbott.co.uk. To confirm the practical reliability, we examined the agreement of experimentally obtained HSP data from the literature for 93 compounds and the data predicted by the created models. The results of GNN modeling showed the best predictive characteristics, which include a coefficient of determination between experimentally obtained and predicted HSP values greater than 0.76 for polar and hydrogen bond forces and greater than 0.66 for dispersive forces. Interpreting the fundamental basis of Hansen solubility using the created MLR equations and XGBoost models, HSP values were found to be influenced by van der Waals volume characteristics, 2D matrix molecular representation, and polarity. We elaborated on the practical benefits of using the selected GNN method through Hansen's solubility sphere as an example. This is the first study to demonstrate the advantages of GNN in predicting individual HSP components, as well as the first study to describe in detail their molecular basis using MLR and XGBoost modeling.
期刊介绍:
Chemometrics and Intelligent Laboratory Systems publishes original research papers, short communications, reviews, tutorials and Original Software Publications reporting on development of novel statistical, mathematical, or computer techniques in Chemistry and related disciplines.
Chemometrics is the chemical discipline that uses mathematical and statistical methods to design or select optimal procedures and experiments, and to provide maximum chemical information by analysing chemical data.
The journal deals with the following topics:
1) Development of new statistical, mathematical and chemometrical methods for Chemistry and related fields (Environmental Chemistry, Biochemistry, Toxicology, System Biology, -Omics, etc.)
2) Novel applications of chemometrics to all branches of Chemistry and related fields (typical domains of interest are: process data analysis, experimental design, data mining, signal processing, supervised modelling, decision making, robust statistics, mixture analysis, multivariate calibration etc.) Routine applications of established chemometrical techniques will not be considered.
3) Development of new software that provides novel tools or truly advances the use of chemometrical methods.
4) Well characterized data sets to test performance for the new methods and software.
The journal complies with International Committee of Medical Journal Editors'' Uniform requirements for manuscripts.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


