Daniel R Tabet, Da Kuang, Megan C Lancaster, Roujia Li, Karen Liu, Jochen Weile, Atina G Coté, Yingzhou Wu, Robert A Hegele, Dan M Roden, Frederick P Roth
{"title":"根据推断人类特征的能力对计算变异效应预测器进行标杆分析。","authors":"Daniel R Tabet, Da Kuang, Megan C Lancaster, Roujia Li, Karen Liu, Jochen Weile, Atina G Coté, Yingzhou Wu, Robert A Hegele, Dan M Roden, Frederick P Roth","doi":"10.1186/s13059-024-03314-7","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Computational variant effect predictors offer a scalable and increasingly reliable means of interpreting human genetic variation, but concerns of circularity and bias have limited previous methods for evaluating and comparing predictors. Population-level cohorts of genotyped and phenotyped participants that have not been used in predictor training can facilitate an unbiased benchmarking of available methods. Using a curated set of human gene-trait associations with a reported rare-variant burden association, we evaluate the correlations of 24 computational variant effect predictors with associated human traits in the UK Biobank and All of Us cohorts.</p><p><strong>Results: </strong>AlphaMissense outperformed all other predictors in inferring human traits based on rare missense variants in UK Biobank and All of Us participants. The overall rankings of computational variant effect predictors in these two cohorts showed a significant positive correlation.</p><p><strong>Conclusion: </strong>We describe a method to assess computational variant effect predictors that sidesteps the limitations of previous evaluations. This approach is generalizable to future predictors and could continue to inform predictor choice for personal and clinical genetics.</p>","PeriodicalId":48922,"journal":{"name":"Genome Biology","volume":"25 1","pages":"172"},"PeriodicalIF":12.3000,"publicationDate":"2024-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11218265/pdf/","citationCount":"0","resultStr":"{\"title\":\"Benchmarking computational variant effect predictors by their ability to infer human traits.\",\"authors\":\"Daniel R Tabet, Da Kuang, Megan C Lancaster, Roujia Li, Karen Liu, Jochen Weile, Atina G Coté, Yingzhou Wu, Robert A Hegele, Dan M Roden, Frederick P Roth\",\"doi\":\"10.1186/s13059-024-03314-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Computational variant effect predictors offer a scalable and increasingly reliable means of interpreting human genetic variation, but concerns of circularity and bias have limited previous methods for evaluating and comparing predictors. Population-level cohorts of genotyped and phenotyped participants that have not been used in predictor training can facilitate an unbiased benchmarking of available methods. Using a curated set of human gene-trait associations with a reported rare-variant burden association, we evaluate the correlations of 24 computational variant effect predictors with associated human traits in the UK Biobank and All of Us cohorts.</p><p><strong>Results: </strong>AlphaMissense outperformed all other predictors in inferring human traits based on rare missense variants in UK Biobank and All of Us participants. The overall rankings of computational variant effect predictors in these two cohorts showed a significant positive correlation.</p><p><strong>Conclusion: </strong>We describe a method to assess computational variant effect predictors that sidesteps the limitations of previous evaluations. This approach is generalizable to future predictors and could continue to inform predictor choice for personal and clinical genetics.</p>\",\"PeriodicalId\":48922,\"journal\":{\"name\":\"Genome Biology\",\"volume\":\"25 1\",\"pages\":\"172\"},\"PeriodicalIF\":12.3000,\"publicationDate\":\"2024-07-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11218265/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Genome Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s13059-024-03314-7\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"Agricultural and Biological Sciences\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genome Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13059-024-03314-7","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Agricultural and Biological Sciences","Score":null,"Total":0}
Benchmarking computational variant effect predictors by their ability to infer human traits.
Background: Computational variant effect predictors offer a scalable and increasingly reliable means of interpreting human genetic variation, but concerns of circularity and bias have limited previous methods for evaluating and comparing predictors. Population-level cohorts of genotyped and phenotyped participants that have not been used in predictor training can facilitate an unbiased benchmarking of available methods. Using a curated set of human gene-trait associations with a reported rare-variant burden association, we evaluate the correlations of 24 computational variant effect predictors with associated human traits in the UK Biobank and All of Us cohorts.
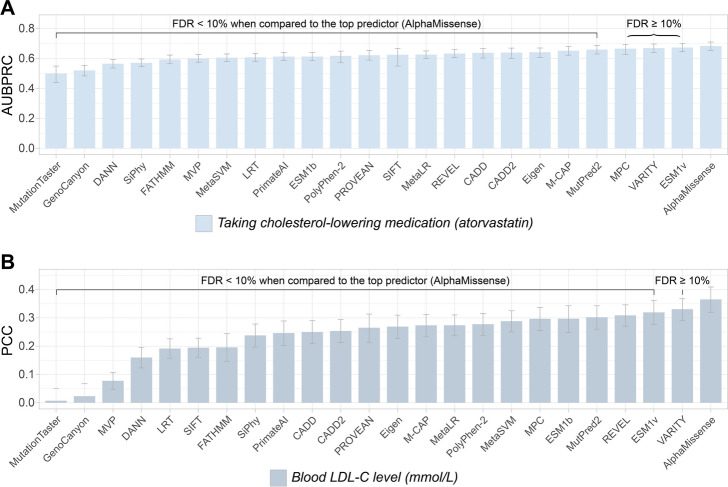
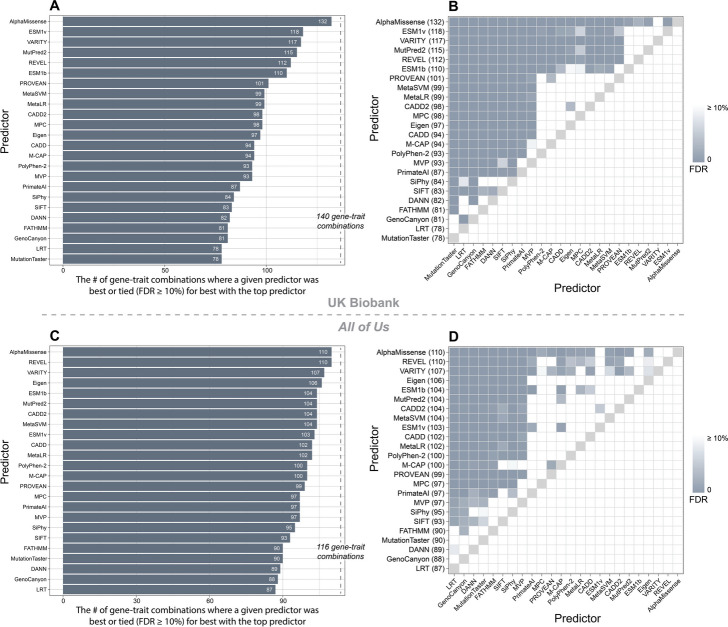
Results: AlphaMissense outperformed all other predictors in inferring human traits based on rare missense variants in UK Biobank and All of Us participants. The overall rankings of computational variant effect predictors in these two cohorts showed a significant positive correlation.
Conclusion: We describe a method to assess computational variant effect predictors that sidesteps the limitations of previous evaluations. This approach is generalizable to future predictors and could continue to inform predictor choice for personal and clinical genetics.
期刊介绍:
Genome Biology is a leading research journal that focuses on the study of biology and biomedicine from a genomic and post-genomic standpoint. The journal consistently publishes outstanding research across various areas within these fields.
With an impressive impact factor of 12.3 (2022), Genome Biology has earned its place as the 3rd highest-ranked research journal in the Genetics and Heredity category, according to Thomson Reuters. Additionally, it is ranked 2nd among research journals in the Biotechnology and Applied Microbiology category. It is important to note that Genome Biology is the top-ranking open access journal in this category.
In summary, Genome Biology sets a high standard for scientific publications in the field, showcasing cutting-edge research and earning recognition among its peers.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


