{"title":"α1-酸糖蛋白 A 变体中 Phe112、Ser114 和 Tyr115 对药物结合选择性的贡献","authors":"Yuka Nakamura, Hiroshi Watanabe, Teruya Nakamura, Mami Chirifu, Kana Ishiodori, Tadashi Imafuku, Hitoshi Maeda, Yoshihiro Kobashigawa, Hiroshi Morioka, Toru Maruyama","doi":"10.1021/acs.molpharmaceut.4c00428","DOIUrl":null,"url":null,"abstract":"<p><p>The plasma protein α<sub>1</sub>-acid glycoprotein (AGP) primarily affects the pharmacokinetics of basic drugs. There are two AGP variants in humans, A and F1*S, exhibiting distinct drug-binding selectivity. Elucidation of the drug-binding selectivity of human AGP variants is essential for drug development and personalized drug therapy. Herein, we aimed to establish the contribution of amino acids 112 and 114 of human AGP to drug-binding selectively. Both amino acids are located in the drug-binding region and differ between the variants. Phe112/Ser114 of the A variant and its equivalent residues in the F1*S variant (Leu112/Phe114) were swapped with each other. Binding experiments were then conducted using the antiarrhythmic drug disopyramide, which selectively binds to the A variant. A significant decrease in the bound fraction was observed in each singly mutated A protein (Phe112Leu or Ser114Phe). Moreover, the bound fraction of the double A mutant (Phe112Leu/Ser114Phe) was decreased to that of wild-type F1*S. Intriguingly, the double F1*S mutant (Leu112Phe/Phe114Ser), in which residues were swapped with those of the A variant, showed only partial restoration in binding. The triple F1*S mutant (Leu112Phe/Phe114Ser/Asp115Tyr), where position 115 is thought to contribute to the difference in pocket size between variants, showed a further recovery in binding to 70% of that of wild-type A. These results were supported by thermodynamic analysis and acridine orange binding, which selectively binds the A variant. Together, these data indicate that, in addition to direct interaction with Phe112 and Ser114, the binding pocket size contributed by Tyr115 is important for the drug-binding selectivity of the A variant.</p>","PeriodicalId":52,"journal":{"name":"Molecular Pharmaceutics","volume":" ","pages":"4038-4046"},"PeriodicalIF":4.5000,"publicationDate":"2024-08-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Contribution of Phe112, Ser114, and Tyr115 to Drug-Binding Selectivity in the A Variant of α<sub>1</sub>-Acid Glycoprotein.\",\"authors\":\"Yuka Nakamura, Hiroshi Watanabe, Teruya Nakamura, Mami Chirifu, Kana Ishiodori, Tadashi Imafuku, Hitoshi Maeda, Yoshihiro Kobashigawa, Hiroshi Morioka, Toru Maruyama\",\"doi\":\"10.1021/acs.molpharmaceut.4c00428\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The plasma protein α<sub>1</sub>-acid glycoprotein (AGP) primarily affects the pharmacokinetics of basic drugs. There are two AGP variants in humans, A and F1*S, exhibiting distinct drug-binding selectivity. Elucidation of the drug-binding selectivity of human AGP variants is essential for drug development and personalized drug therapy. Herein, we aimed to establish the contribution of amino acids 112 and 114 of human AGP to drug-binding selectively. Both amino acids are located in the drug-binding region and differ between the variants. Phe112/Ser114 of the A variant and its equivalent residues in the F1*S variant (Leu112/Phe114) were swapped with each other. Binding experiments were then conducted using the antiarrhythmic drug disopyramide, which selectively binds to the A variant. A significant decrease in the bound fraction was observed in each singly mutated A protein (Phe112Leu or Ser114Phe). Moreover, the bound fraction of the double A mutant (Phe112Leu/Ser114Phe) was decreased to that of wild-type F1*S. Intriguingly, the double F1*S mutant (Leu112Phe/Phe114Ser), in which residues were swapped with those of the A variant, showed only partial restoration in binding. The triple F1*S mutant (Leu112Phe/Phe114Ser/Asp115Tyr), where position 115 is thought to contribute to the difference in pocket size between variants, showed a further recovery in binding to 70% of that of wild-type A. These results were supported by thermodynamic analysis and acridine orange binding, which selectively binds the A variant. Together, these data indicate that, in addition to direct interaction with Phe112 and Ser114, the binding pocket size contributed by Tyr115 is important for the drug-binding selectivity of the A variant.</p>\",\"PeriodicalId\":52,\"journal\":{\"name\":\"Molecular Pharmaceutics\",\"volume\":\" \",\"pages\":\"4038-4046\"},\"PeriodicalIF\":4.5000,\"publicationDate\":\"2024-08-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Pharmaceutics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.molpharmaceut.4c00428\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/7/1 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"MEDICINE, RESEARCH & EXPERIMENTAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Pharmaceutics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1021/acs.molpharmaceut.4c00428","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/1 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
引用次数: 0
摘要
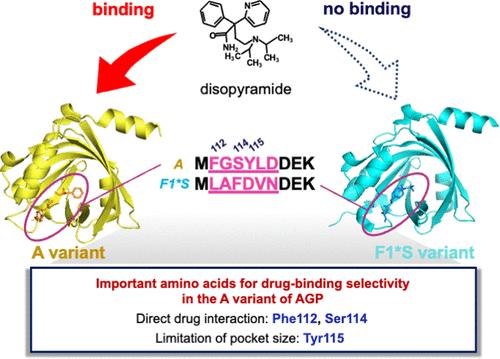
血浆蛋白α1-酸性糖蛋白(AGP)主要影响基本药物的药代动力学。人类有两种 AGP 变体:A 和 F1*S,它们表现出不同的药物结合选择性。阐明人类 AGP 变体的药物结合选择性对于药物开发和个性化药物治疗至关重要。在此,我们旨在确定人类 AGP 的 112 和 114 氨基酸对药物结合选择性的贡献。这两个氨基酸都位于药物结合区,并且在不同变体之间存在差异。我们将 A 变体中的 Phe112/Ser114 与 F1*S 变体中的等效残基(Leu112/Phe114)互换。然后使用选择性与 A 变体结合的抗心律失常药物地氯雷他定进行了结合实验。结果发现,单突变 A 蛋白(Phe112Leu 或 Ser114Phe)的结合率明显下降。此外,双 A 突变体(Phe112Leu/Ser114Phe)的结合率也下降到野生型 F1*S 的水平。耐人寻味的是,双 F1*S 突变体(Leu112Phe/Phe114Ser)中的残基与 A 突变体中的残基互换后,结合率仅部分恢复。三重 F1*S 突变体(Leu112Phe/Phe114Ser/Asp115Tyr)显示,其结合力进一步恢复到野生型 A 的 70%,而热力学分析和吖啶橙结合(可选择性结合 A 变体)均支持这些结果。这些数据共同表明,除了与 Phe112 和 Ser114 直接相互作用外,Tyr115 的结合口袋大小对 A 变体的药物结合选择性也很重要。
Contribution of Phe112, Ser114, and Tyr115 to Drug-Binding Selectivity in the A Variant of α1-Acid Glycoprotein.
The plasma protein α1-acid glycoprotein (AGP) primarily affects the pharmacokinetics of basic drugs. There are two AGP variants in humans, A and F1*S, exhibiting distinct drug-binding selectivity. Elucidation of the drug-binding selectivity of human AGP variants is essential for drug development and personalized drug therapy. Herein, we aimed to establish the contribution of amino acids 112 and 114 of human AGP to drug-binding selectively. Both amino acids are located in the drug-binding region and differ between the variants. Phe112/Ser114 of the A variant and its equivalent residues in the F1*S variant (Leu112/Phe114) were swapped with each other. Binding experiments were then conducted using the antiarrhythmic drug disopyramide, which selectively binds to the A variant. A significant decrease in the bound fraction was observed in each singly mutated A protein (Phe112Leu or Ser114Phe). Moreover, the bound fraction of the double A mutant (Phe112Leu/Ser114Phe) was decreased to that of wild-type F1*S. Intriguingly, the double F1*S mutant (Leu112Phe/Phe114Ser), in which residues were swapped with those of the A variant, showed only partial restoration in binding. The triple F1*S mutant (Leu112Phe/Phe114Ser/Asp115Tyr), where position 115 is thought to contribute to the difference in pocket size between variants, showed a further recovery in binding to 70% of that of wild-type A. These results were supported by thermodynamic analysis and acridine orange binding, which selectively binds the A variant. Together, these data indicate that, in addition to direct interaction with Phe112 and Ser114, the binding pocket size contributed by Tyr115 is important for the drug-binding selectivity of the A variant.
期刊介绍:
Molecular Pharmaceutics publishes the results of original research that contributes significantly to the molecular mechanistic understanding of drug delivery and drug delivery systems. The journal encourages contributions describing research at the interface of drug discovery and drug development.
Scientific areas within the scope of the journal include physical and pharmaceutical chemistry, biochemistry and biophysics, molecular and cellular biology, and polymer and materials science as they relate to drug and drug delivery system efficacy. Mechanistic Drug Delivery and Drug Targeting research on modulating activity and efficacy of a drug or drug product is within the scope of Molecular Pharmaceutics. Theoretical and experimental peer-reviewed research articles, communications, reviews, and perspectives are welcomed.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


