利用分子特征表示和机器学习预测真空紫外/紫外气相吸收光谱
IF 5.6
2区 化学
Q1 CHEMISTRY, MEDICINAL
引用次数: 0
摘要
紫外线(UV)吸收光谱是一种广泛用于定量和定性分析化合物的工具。在气相中,真空紫外(VUV)和紫外吸收光谱对许多小分子具有特异性和诊断性。准确预测真空紫外/紫外吸收光谱有助于鉴定燃料、法医和药物研究等领域的新分子或未知分子。量子化学光谱预测的另一种方法是使用人工智能。在此,我们使用并开发了不同的分子特征表示技术来编码化学结构,以测试预测气相紫外/紫外吸收光谱的三种机器学习模型。1397 种挥发性和半挥发性化合物的结构数据文件(.sdf)和紫外/紫外吸收光谱被用来训练和测试模型。引入了新的分子特征(称为 ABOCH),以更好地捕捉π键、芳香性和卤化。与计算密集型的基于分子的深度学习方法相比,这些新特征的加入有利于光谱预测,并表现出更优越的性能。在机器学习方法中,使用随机森林回归器的准确率最高,训练时间最短。所开发的机器学习预测模型也优于基于时变密度泛函理论的光谱预测。本文章由计算机程序翻译,如有差异,请以英文原文为准。
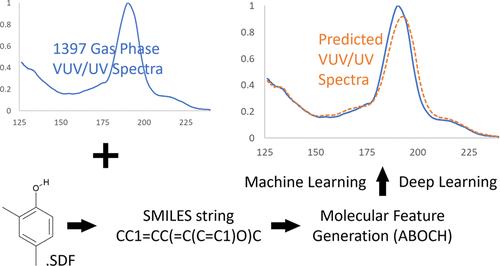
Prediction of Vacuum Ultraviolet/Ultraviolet Gas-Phase Absorption Spectra Using Molecular Feature Representations and Machine Learning
Ultraviolet (UV) absorption spectroscopy is a widely used tool for quantitative and qualitative analyses of chemical compounds. In the gas phase, vacuum UV (VUV) and UV absorption spectra are specific and diagnostic for many small molecules. An accurate prediction of VUV/UV absorption spectra can aid the characterization of new or unknown molecules in areas such as fuels, forensics, and pharmaceutical research. An alternative to quantum chemical spectral prediction is the use of artificial intelligence. Here, different molecular feature representation techniques were used and developed to encode chemical structures for testing three machine learning models to predict gas-phase VUV/UV absorption spectra. Structure data files (.sdf) and VUV/UV absorption spectra for 1397 volatile and semivolatile chemical compounds were used to train and test the models. New molecular features (termed ABOCH) were introduced to better capture pi-bonding, aromaticity, and halogenation. The incorporation of these new features benefited spectral prediction and demonstrated superior performance compared to computationally intensive molecular-based deep learning methods. Of the machine learning methods, the use of a Random Forest regressor returned the best accuracy score with the shortest training time. The developed machine learning prediction model also outperformed spectral predictions based on the time-dependent density functional theory.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

CiteScore
9.80
自引率
10.70%
发文量
529
审稿时长
1.4 months
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


