Ziquan Zeng, Jianchuan Wang, Shiwei Zhang, Bo Han, Feng Dang, Songlin Li and Yong Du
{"title":"了解掺镁 Li2MnO3 的电化学特性:第一原理计算。","authors":"Ziquan Zeng, Jianchuan Wang, Shiwei Zhang, Bo Han, Feng Dang, Songlin Li and Yong Du","doi":"10.1039/D4CP01733A","DOIUrl":null,"url":null,"abstract":"<p >Non-transition metal doping, especially for Mg, has been gradually employed to optimize the electrochemical performance of Li-rich cathode material Li<small><sub>2</sub></small>MnO<small><sub>3</sub></small>. However, the effects of Mg doping on the electrochemical behavior of Li<small><sub>2</sub></small>MnO<small><sub>3</sub></small> have not been studied extensively. In this work, we investigate the effect of Mg doping at both the 2b (in the Li/Mn mixed layer) and 4h (in the Li layer) Li sites on the electrochemical properties of Li<small><sub>2</sub></small>MnO<small><sub>3</sub></small> through first-principles calculations and <em>ab initi</em>o molecular dynamics simulations. The local lattice structure, electronic density of states, Bader charge, delithiation voltage, lattice oxygen stability and Li diffusion kinetics are examined. Electronic structure analysis shows that Mg can activate the electrochemical activity of surrounding Mn by charge transfer, making Mn participate in charge compensation at the initial delithiation stage. Mg doping can also cause an increase in the average oxygen vacancy formation energy and hence depress the oxygen release during the delithiation process. Molecular dynamics simulations show that the diffusion kinetics of Li ions in Mg<small><sub>2b</sub></small>–Li<small><sub>2</sub></small>MnO<small><sub>3</sub></small> is enhanced with respect to the undoped one, whereas Mg doped at the 4h site cannot improve the diffusion kinetics of Li ions. Further studies found that Mg doped at the 2b site results in a decrease in the energy barrier for the intra-layer diffusion and an increase in the energy barrier for the inter-layer diffusion of the nearby Li vacancies.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":null,"pages":null},"PeriodicalIF":2.9000,"publicationDate":"2024-06-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Understanding the electrochemical properties of Mg-doped Li2MnO3: first-principles calculations†\",\"authors\":\"Ziquan Zeng, Jianchuan Wang, Shiwei Zhang, Bo Han, Feng Dang, Songlin Li and Yong Du\",\"doi\":\"10.1039/D4CP01733A\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Non-transition metal doping, especially for Mg, has been gradually employed to optimize the electrochemical performance of Li-rich cathode material Li<small><sub>2</sub></small>MnO<small><sub>3</sub></small>. However, the effects of Mg doping on the electrochemical behavior of Li<small><sub>2</sub></small>MnO<small><sub>3</sub></small> have not been studied extensively. In this work, we investigate the effect of Mg doping at both the 2b (in the Li/Mn mixed layer) and 4h (in the Li layer) Li sites on the electrochemical properties of Li<small><sub>2</sub></small>MnO<small><sub>3</sub></small> through first-principles calculations and <em>ab initi</em>o molecular dynamics simulations. The local lattice structure, electronic density of states, Bader charge, delithiation voltage, lattice oxygen stability and Li diffusion kinetics are examined. Electronic structure analysis shows that Mg can activate the electrochemical activity of surrounding Mn by charge transfer, making Mn participate in charge compensation at the initial delithiation stage. Mg doping can also cause an increase in the average oxygen vacancy formation energy and hence depress the oxygen release during the delithiation process. Molecular dynamics simulations show that the diffusion kinetics of Li ions in Mg<small><sub>2b</sub></small>–Li<small><sub>2</sub></small>MnO<small><sub>3</sub></small> is enhanced with respect to the undoped one, whereas Mg doped at the 4h site cannot improve the diffusion kinetics of Li ions. Further studies found that Mg doped at the 2b site results in a decrease in the energy barrier for the intra-layer diffusion and an increase in the energy barrier for the inter-layer diffusion of the nearby Li vacancies.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2024-06-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2024/cp/d4cp01733a\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/cp/d4cp01733a","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Understanding the electrochemical properties of Mg-doped Li2MnO3: first-principles calculations†
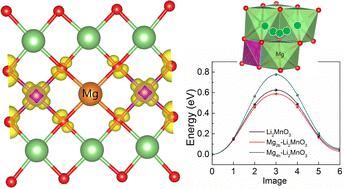
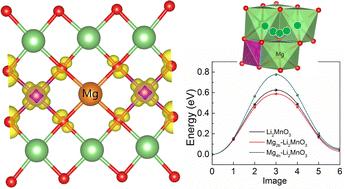
Non-transition metal doping, especially for Mg, has been gradually employed to optimize the electrochemical performance of Li-rich cathode material Li2MnO3. However, the effects of Mg doping on the electrochemical behavior of Li2MnO3 have not been studied extensively. In this work, we investigate the effect of Mg doping at both the 2b (in the Li/Mn mixed layer) and 4h (in the Li layer) Li sites on the electrochemical properties of Li2MnO3 through first-principles calculations and ab initio molecular dynamics simulations. The local lattice structure, electronic density of states, Bader charge, delithiation voltage, lattice oxygen stability and Li diffusion kinetics are examined. Electronic structure analysis shows that Mg can activate the electrochemical activity of surrounding Mn by charge transfer, making Mn participate in charge compensation at the initial delithiation stage. Mg doping can also cause an increase in the average oxygen vacancy formation energy and hence depress the oxygen release during the delithiation process. Molecular dynamics simulations show that the diffusion kinetics of Li ions in Mg2b–Li2MnO3 is enhanced with respect to the undoped one, whereas Mg doped at the 4h site cannot improve the diffusion kinetics of Li ions. Further studies found that Mg doped at the 2b site results in a decrease in the energy barrier for the intra-layer diffusion and an increase in the energy barrier for the inter-layer diffusion of the nearby Li vacancies.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


