Francesco Muniz-Miranda, Alfonso Pedone, Maria Cristina Menziani
{"title":"溶液中乙腈 CN 伸展振动的蓝移:计算和实验研究","authors":"Francesco Muniz-Miranda, Alfonso Pedone, Maria Cristina Menziani","doi":"10.1002/jcc.27452","DOIUrl":null,"url":null,"abstract":"<p>Acetonitrile, a polar molecule that cannot form hydrogen bonds on its own, interacts with solvent molecules mainly through the lone pair of its nitrogen atom and the π electrons of its CN triple bond [Correction added on 17 July 2024, after first online publication: Acetole has been changed to Acetonitrile in the preceeding sentence.]. Interestingly, acetonitrile exhibits an unexpected strengthening of the triple bond's force constant in an aqueous environment, leading to an upshift (<i>blueshift</i>) in the corresponding stretching vibration: this effect contrasts with the usual consequence of hydrogen bonding on the vibrational frequencies of the acceptor groups, that is, frequency <i>redshift</i>. This investigation elucidates this phenomenon using Raman spectroscopy to examine the behavior of acetonitrile in organic solvent, water, and silver ion aqueous solutions, where an even more pronounced upshift is observed. Raman spectroscopy is particularly well suited for analyzing aqueous solutions due to the minimal scattering effect of water molecules across most of the vibrational spectrum. Computational approaches, both static and dynamical, based on Density Functional Theory and hybrid functionals, are employed here to interpret these findings, and accurately reproduce the vibrational frequencies of acetonitrile in different environments. Our calculations also allow an explanation for this unique behavior in terms of electric charge displacements. On the other hand, the study of the interaction of acetonitrile with water molecules and metal ions is relevant for the use of this molecule as a solvent in both chemical and pharmaceutical applications.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 28","pages":"2352-2359"},"PeriodicalIF":3.4000,"publicationDate":"2024-06-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Blueshift of the CN stretching vibration of acetonitrile in solution: computational and experimental study\",\"authors\":\"Francesco Muniz-Miranda, Alfonso Pedone, Maria Cristina Menziani\",\"doi\":\"10.1002/jcc.27452\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Acetonitrile, a polar molecule that cannot form hydrogen bonds on its own, interacts with solvent molecules mainly through the lone pair of its nitrogen atom and the π electrons of its CN triple bond [Correction added on 17 July 2024, after first online publication: Acetole has been changed to Acetonitrile in the preceeding sentence.]. Interestingly, acetonitrile exhibits an unexpected strengthening of the triple bond's force constant in an aqueous environment, leading to an upshift (<i>blueshift</i>) in the corresponding stretching vibration: this effect contrasts with the usual consequence of hydrogen bonding on the vibrational frequencies of the acceptor groups, that is, frequency <i>redshift</i>. This investigation elucidates this phenomenon using Raman spectroscopy to examine the behavior of acetonitrile in organic solvent, water, and silver ion aqueous solutions, where an even more pronounced upshift is observed. Raman spectroscopy is particularly well suited for analyzing aqueous solutions due to the minimal scattering effect of water molecules across most of the vibrational spectrum. Computational approaches, both static and dynamical, based on Density Functional Theory and hybrid functionals, are employed here to interpret these findings, and accurately reproduce the vibrational frequencies of acetonitrile in different environments. Our calculations also allow an explanation for this unique behavior in terms of electric charge displacements. On the other hand, the study of the interaction of acetonitrile with water molecules and metal ions is relevant for the use of this molecule as a solvent in both chemical and pharmaceutical applications.</p>\",\"PeriodicalId\":188,\"journal\":{\"name\":\"Journal of Computational Chemistry\",\"volume\":\"45 28\",\"pages\":\"2352-2359\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2024-06-20\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computational Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27452\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27452","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Blueshift of the CN stretching vibration of acetonitrile in solution: computational and experimental study
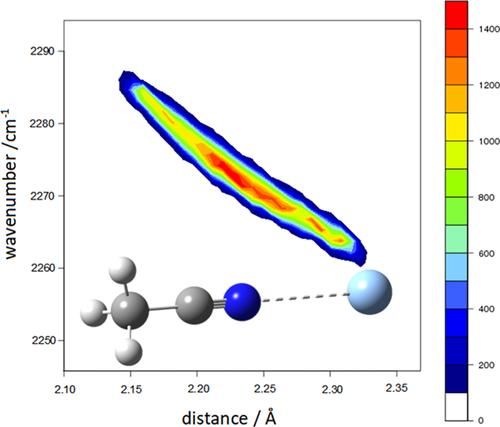
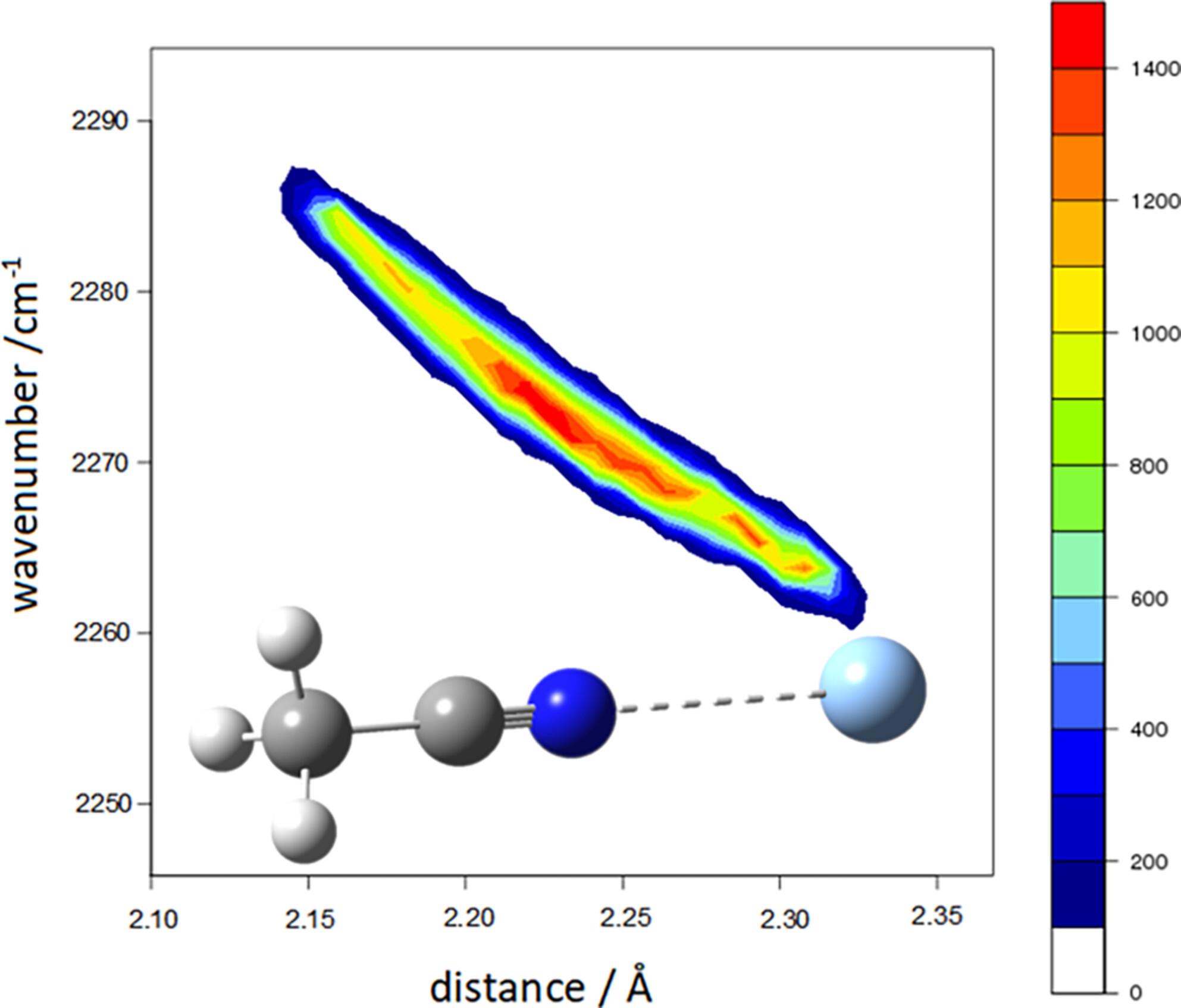
Acetonitrile, a polar molecule that cannot form hydrogen bonds on its own, interacts with solvent molecules mainly through the lone pair of its nitrogen atom and the π electrons of its CN triple bond [Correction added on 17 July 2024, after first online publication: Acetole has been changed to Acetonitrile in the preceeding sentence.]. Interestingly, acetonitrile exhibits an unexpected strengthening of the triple bond's force constant in an aqueous environment, leading to an upshift (blueshift) in the corresponding stretching vibration: this effect contrasts with the usual consequence of hydrogen bonding on the vibrational frequencies of the acceptor groups, that is, frequency redshift. This investigation elucidates this phenomenon using Raman spectroscopy to examine the behavior of acetonitrile in organic solvent, water, and silver ion aqueous solutions, where an even more pronounced upshift is observed. Raman spectroscopy is particularly well suited for analyzing aqueous solutions due to the minimal scattering effect of water molecules across most of the vibrational spectrum. Computational approaches, both static and dynamical, based on Density Functional Theory and hybrid functionals, are employed here to interpret these findings, and accurately reproduce the vibrational frequencies of acetonitrile in different environments. Our calculations also allow an explanation for this unique behavior in terms of electric charge displacements. On the other hand, the study of the interaction of acetonitrile with water molecules and metal ions is relevant for the use of this molecule as a solvent in both chemical and pharmaceutical applications.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


