Nickolas A. Joyner, João Gabriel Farias Romeu, Brian Kent and David A. Dixon
{"title":"二原子氧化镍的电子结构","authors":"Nickolas A. Joyner, João Gabriel Farias Romeu, Brian Kent and David A. Dixon","doi":"10.1039/D4CP01796J","DOIUrl":null,"url":null,"abstract":"<p >The nature of the Ni–O bond is relevant to catalytic and environmental applications. The vibrational frequency and electronic structure of NiO were calculated using CASSCF, icMRCI+Q, CCSD(T), and DFT. CASSCF predicted a quintet state (<small><sup>5</sup></small>Σ<small><sup>−</sup></small>) ground state for the equilibrium bond distance with a state crossing at 1.65 Å, where the triplet (<small><sup>3</sup></small>Σ<small><sup>−</sup></small>) state becomes of lower energy. These states arise from the 3d<small><sup>8</sup></small>(<small><sup>3</sup></small>F)4s<small><sup>2</sup></small> (<small><sup>3</sup></small>F) and 3d<small><sup>9</sup></small>(<small><sup>2</sup></small>D)4s<small><sup>1</sup></small> (<small><sup>3</sup></small>D) configurations of Ni. The icMRCI+Q method predicts a triplet (<small><sup>3</sup></small>Σ<small><sup>−</sup></small>) ground state and does not predict a state crossing with the quintet. This state has significant ionic character with the 2p<small><sub><em>z</em></sub></small> of O bonding with the 4s/3d<small><sub><em>z</em><small><sup>2</sup></small></sub></small> of the Ni to form a σ bond. The NiO frequency at the icMRCI+Q level of 835.0 cm<small><sup>−1</sup></small> is in excellent agreement with experiment; the value of <em>r</em><small><sub>e</sub></small> is 1.5992 Å at this computational level. CCSD(T) predicts <em>ω</em><small><sub>e</sub></small> = 888.80 cm<small><sup>−1</sup></small> when extrapolated to the complete basis set limit. Frequencies predicted using CCSD(T) deviate from experiment consistent with the calculations showing large multireference character. A wide array of density functionals were benchmarked. Of the 43 functionals tested, the ones that gave the best prediction of the frequency are ωB97XD, CAM-B3LYP, and τ-HCTH with respective values of 831.8, 838.3, and 837.4 cm<small><sup>−1</sup></small> respectively. The bond dissociation energy (BDE) of NiO is predicted to be 352.4 kJ mol<small><sup>−1</sup></small> at the Feller–Peterson–Dixon (FPD) level in good agreement with one of the experimental values. The calculated BDEs at the DFT level are sensitive to the choice of functional and atomic asymptote. Sixteen functionals predicted the BDE within 20 kJ mol<small><sup>−1</sup></small> of the FPD value.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 29","pages":" 19646-19657"},"PeriodicalIF":2.9000,"publicationDate":"2024-06-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"The electronic structure of diatomic nickel oxide†\",\"authors\":\"Nickolas A. Joyner, João Gabriel Farias Romeu, Brian Kent and David A. Dixon\",\"doi\":\"10.1039/D4CP01796J\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The nature of the Ni–O bond is relevant to catalytic and environmental applications. The vibrational frequency and electronic structure of NiO were calculated using CASSCF, icMRCI+Q, CCSD(T), and DFT. CASSCF predicted a quintet state (<small><sup>5</sup></small>Σ<small><sup>−</sup></small>) ground state for the equilibrium bond distance with a state crossing at 1.65 Å, where the triplet (<small><sup>3</sup></small>Σ<small><sup>−</sup></small>) state becomes of lower energy. These states arise from the 3d<small><sup>8</sup></small>(<small><sup>3</sup></small>F)4s<small><sup>2</sup></small> (<small><sup>3</sup></small>F) and 3d<small><sup>9</sup></small>(<small><sup>2</sup></small>D)4s<small><sup>1</sup></small> (<small><sup>3</sup></small>D) configurations of Ni. The icMRCI+Q method predicts a triplet (<small><sup>3</sup></small>Σ<small><sup>−</sup></small>) ground state and does not predict a state crossing with the quintet. This state has significant ionic character with the 2p<small><sub><em>z</em></sub></small> of O bonding with the 4s/3d<small><sub><em>z</em><small><sup>2</sup></small></sub></small> of the Ni to form a σ bond. The NiO frequency at the icMRCI+Q level of 835.0 cm<small><sup>−1</sup></small> is in excellent agreement with experiment; the value of <em>r</em><small><sub>e</sub></small> is 1.5992 Å at this computational level. CCSD(T) predicts <em>ω</em><small><sub>e</sub></small> = 888.80 cm<small><sup>−1</sup></small> when extrapolated to the complete basis set limit. Frequencies predicted using CCSD(T) deviate from experiment consistent with the calculations showing large multireference character. A wide array of density functionals were benchmarked. Of the 43 functionals tested, the ones that gave the best prediction of the frequency are ωB97XD, CAM-B3LYP, and τ-HCTH with respective values of 831.8, 838.3, and 837.4 cm<small><sup>−1</sup></small> respectively. The bond dissociation energy (BDE) of NiO is predicted to be 352.4 kJ mol<small><sup>−1</sup></small> at the Feller–Peterson–Dixon (FPD) level in good agreement with one of the experimental values. The calculated BDEs at the DFT level are sensitive to the choice of functional and atomic asymptote. Sixteen functionals predicted the BDE within 20 kJ mol<small><sup>−1</sup></small> of the FPD value.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 29\",\"pages\":\" 19646-19657\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2024-06-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2024/cp/d4cp01796j\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/cp/d4cp01796j","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
The electronic structure of diatomic nickel oxide†
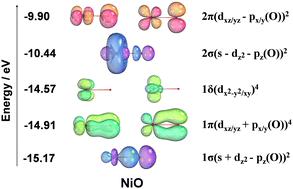
The nature of the Ni–O bond is relevant to catalytic and environmental applications. The vibrational frequency and electronic structure of NiO were calculated using CASSCF, icMRCI+Q, CCSD(T), and DFT. CASSCF predicted a quintet state (5Σ−) ground state for the equilibrium bond distance with a state crossing at 1.65 Å, where the triplet (3Σ−) state becomes of lower energy. These states arise from the 3d8(3F)4s2 (3F) and 3d9(2D)4s1 (3D) configurations of Ni. The icMRCI+Q method predicts a triplet (3Σ−) ground state and does not predict a state crossing with the quintet. This state has significant ionic character with the 2pz of O bonding with the 4s/3dz2 of the Ni to form a σ bond. The NiO frequency at the icMRCI+Q level of 835.0 cm−1 is in excellent agreement with experiment; the value of re is 1.5992 Å at this computational level. CCSD(T) predicts ωe = 888.80 cm−1 when extrapolated to the complete basis set limit. Frequencies predicted using CCSD(T) deviate from experiment consistent with the calculations showing large multireference character. A wide array of density functionals were benchmarked. Of the 43 functionals tested, the ones that gave the best prediction of the frequency are ωB97XD, CAM-B3LYP, and τ-HCTH with respective values of 831.8, 838.3, and 837.4 cm−1 respectively. The bond dissociation energy (BDE) of NiO is predicted to be 352.4 kJ mol−1 at the Feller–Peterson–Dixon (FPD) level in good agreement with one of the experimental values. The calculated BDEs at the DFT level are sensitive to the choice of functional and atomic asymptote. Sixteen functionals predicted the BDE within 20 kJ mol−1 of the FPD value.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


