{"title":"18q缺失综合征表现为迟发型联合免疫缺陷症","authors":"Sho Hashiguchi, Dan Tomomasa, Takuro Nishikawa, Shuji Ishikawa, Harumi Akaike, Hidehiko Kobae, Tsuyoshi Shirai, Toshikage Nagao, Kosuke Noma, Satoshi Okada, Kazuhiro Kamuro, Yasuhiro Okamoto, Hirokazu Kanegane","doi":"10.1007/s10875-024-01751-4","DOIUrl":null,"url":null,"abstract":"<p><p>Patients with chromosome 18q deletion syndrome generally experience hypogammaglobulinemia. Herein, we describe two patients with chromosome 18q deletion syndrome who presented with late-onset combined immune deficiency (LOCID), which has not been previously reported. Patient 1 was a 29-year-old male with 18q deletion syndrome, who was being managed for severe motor and intellectual disabilities at the Yamabiko Medical Welfare Center for 26 years. Although the patient had few infections, he developed Pneumocystis pneumonia at the age of 28. Patient 2, a 48-year-old female with intellectual disability and congenital malformations, was referred to Tokyo Medical and Dental University Hospital with abnormal bilateral lung shadows detected on her chest radiography. Computed tomography showed multiple lymphadenopathies and pneumonia. A lymph node biopsy of the inguinal region revealed granulomatous lymphadenitis, and a chromosomal examination revealed 18q deletion. Array-based genomic hybridization analysis revealed deletion at 18q21.32-q22.3 for patient 1 and at 18q21.33-qter for patient 2. Immune status work-up of the two patients revealed panhypogammaglobulinemia, decreased number of memory B cells and naïve CD4<sup>+</sup> and/or CD8<sup>+</sup> cells, reduced response on the carboxyfluorescein diacetate succinimidyl ester T-cell division test, and low levels of T-cell receptor recombination excision circles and Ig κ-deleting recombination excision circles. Consequently, both patients were diagnosed with LOCID. Although patients with 18q deletion syndrome generally experience humoral immunodeficiency, the disease can be further complicated by cell-mediated immunodeficiency, causing combined immunodeficiency. Therefore, patients with 18q deletion syndrome should be regularly tested for cellular/humoral immunocompetence.</p>","PeriodicalId":15531,"journal":{"name":"Journal of Clinical Immunology","volume":"44 7","pages":"154"},"PeriodicalIF":7.2000,"publicationDate":"2024-06-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11186878/pdf/","citationCount":"0","resultStr":"{\"title\":\"18q Deletion Syndrome Presenting with Late-Onset Combined Immunodeficiency.\",\"authors\":\"Sho Hashiguchi, Dan Tomomasa, Takuro Nishikawa, Shuji Ishikawa, Harumi Akaike, Hidehiko Kobae, Tsuyoshi Shirai, Toshikage Nagao, Kosuke Noma, Satoshi Okada, Kazuhiro Kamuro, Yasuhiro Okamoto, Hirokazu Kanegane\",\"doi\":\"10.1007/s10875-024-01751-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Patients with chromosome 18q deletion syndrome generally experience hypogammaglobulinemia. Herein, we describe two patients with chromosome 18q deletion syndrome who presented with late-onset combined immune deficiency (LOCID), which has not been previously reported. Patient 1 was a 29-year-old male with 18q deletion syndrome, who was being managed for severe motor and intellectual disabilities at the Yamabiko Medical Welfare Center for 26 years. Although the patient had few infections, he developed Pneumocystis pneumonia at the age of 28. Patient 2, a 48-year-old female with intellectual disability and congenital malformations, was referred to Tokyo Medical and Dental University Hospital with abnormal bilateral lung shadows detected on her chest radiography. Computed tomography showed multiple lymphadenopathies and pneumonia. A lymph node biopsy of the inguinal region revealed granulomatous lymphadenitis, and a chromosomal examination revealed 18q deletion. Array-based genomic hybridization analysis revealed deletion at 18q21.32-q22.3 for patient 1 and at 18q21.33-qter for patient 2. Immune status work-up of the two patients revealed panhypogammaglobulinemia, decreased number of memory B cells and naïve CD4<sup>+</sup> and/or CD8<sup>+</sup> cells, reduced response on the carboxyfluorescein diacetate succinimidyl ester T-cell division test, and low levels of T-cell receptor recombination excision circles and Ig κ-deleting recombination excision circles. Consequently, both patients were diagnosed with LOCID. Although patients with 18q deletion syndrome generally experience humoral immunodeficiency, the disease can be further complicated by cell-mediated immunodeficiency, causing combined immunodeficiency. Therefore, patients with 18q deletion syndrome should be regularly tested for cellular/humoral immunocompetence.</p>\",\"PeriodicalId\":15531,\"journal\":{\"name\":\"Journal of Clinical Immunology\",\"volume\":\"44 7\",\"pages\":\"154\"},\"PeriodicalIF\":7.2000,\"publicationDate\":\"2024-06-19\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11186878/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Clinical Immunology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s10875-024-01751-4\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"IMMUNOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Clinical Immunology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10875-024-01751-4","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
18q Deletion Syndrome Presenting with Late-Onset Combined Immunodeficiency.
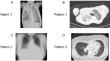
Patients with chromosome 18q deletion syndrome generally experience hypogammaglobulinemia. Herein, we describe two patients with chromosome 18q deletion syndrome who presented with late-onset combined immune deficiency (LOCID), which has not been previously reported. Patient 1 was a 29-year-old male with 18q deletion syndrome, who was being managed for severe motor and intellectual disabilities at the Yamabiko Medical Welfare Center for 26 years. Although the patient had few infections, he developed Pneumocystis pneumonia at the age of 28. Patient 2, a 48-year-old female with intellectual disability and congenital malformations, was referred to Tokyo Medical and Dental University Hospital with abnormal bilateral lung shadows detected on her chest radiography. Computed tomography showed multiple lymphadenopathies and pneumonia. A lymph node biopsy of the inguinal region revealed granulomatous lymphadenitis, and a chromosomal examination revealed 18q deletion. Array-based genomic hybridization analysis revealed deletion at 18q21.32-q22.3 for patient 1 and at 18q21.33-qter for patient 2. Immune status work-up of the two patients revealed panhypogammaglobulinemia, decreased number of memory B cells and naïve CD4+ and/or CD8+ cells, reduced response on the carboxyfluorescein diacetate succinimidyl ester T-cell division test, and low levels of T-cell receptor recombination excision circles and Ig κ-deleting recombination excision circles. Consequently, both patients were diagnosed with LOCID. Although patients with 18q deletion syndrome generally experience humoral immunodeficiency, the disease can be further complicated by cell-mediated immunodeficiency, causing combined immunodeficiency. Therefore, patients with 18q deletion syndrome should be regularly tested for cellular/humoral immunocompetence.
期刊介绍:
The Journal of Clinical Immunology publishes impactful papers in the realm of human immunology, delving into the diagnosis, pathogenesis, prognosis, or treatment of human diseases. The journal places particular emphasis on primary immunodeficiencies and related diseases, encompassing inborn errors of immunity in a broad sense, their underlying genotypes, and diverse phenotypes. These phenotypes include infection, malignancy, allergy, auto-inflammation, and autoimmunity. We welcome a broad spectrum of studies in this domain, spanning genetic discovery, clinical description, immunologic assessment, diagnostic approaches, prognosis evaluation, and treatment interventions. Case reports are considered if they are genuinely original and accompanied by a concise review of the relevant medical literature, illustrating how the novel case study advances the field. The instructions to authors provide detailed guidance on the four categories of papers accepted by the journal.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


