{"title":"GTDrift:用于探索真核生物中遗传漂变、基因组和转录组特征之间相互作用的资源。","authors":"Florian Bénitière, Laurent Duret, Anamaria Necsulea","doi":"10.1093/nargab/lqae064","DOIUrl":null,"url":null,"abstract":"<p><p>We present GTDrift, a comprehensive data resource that enables explorations of genomic and transcriptomic characteristics alongside proxies of the intensity of genetic drift in individual species. This resource encompasses data for 1506 eukaryotic species, including 1413 animals and 93 green plants, and is organized in three components. The first two components contain approximations of the effective population size, which serve as indicators of the extent of random genetic drift within each species. In the first component, we meticulously investigated public databases to assemble data on life history traits such as longevity, adult body length and body mass for a set of 979 species. The second component includes estimations of the ratio between the rate of non-synonymous substitutions and the rate of synonymous substitutions (d<i>N</i>/d<i>S</i>) in protein-coding sequences for 1324 species. This ratio provides an estimate of the efficiency of natural selection in purging deleterious substitutions. Additionally, we present polymorphism-derived <i>N</i> <sub>e</sub> estimates for 66 species. The third component encompasses various genomic and transcriptomic characteristics. With this component, we aim to facilitate comparative transcriptomics analyses across species, by providing easy-to-use processed data for more than 16 000 RNA-seq samples across 491 species. These data include intron-centered alternative splicing frequencies, gene expression levels and sequencing depth statistics for each species, obtained with a homogeneous analysis protocol. To enable cross-species comparisons, we provide orthology predictions for conserved single-copy genes based on BUSCO gene sets. To illustrate the possible uses of this database, we identify the most frequently used introns for each gene and we assess how the sequencing depth available for each species affects our power to identify major and minor splice variants.</p>","PeriodicalId":33994,"journal":{"name":"NAR Genomics and Bioinformatics","volume":"6 2","pages":"lqae064"},"PeriodicalIF":2.8000,"publicationDate":"2024-06-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11167491/pdf/","citationCount":"0","resultStr":"{\"title\":\"GTDrift: a resource for exploring the interplay between genetic drift, genomic and transcriptomic characteristics in eukaryotes.\",\"authors\":\"Florian Bénitière, Laurent Duret, Anamaria Necsulea\",\"doi\":\"10.1093/nargab/lqae064\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>We present GTDrift, a comprehensive data resource that enables explorations of genomic and transcriptomic characteristics alongside proxies of the intensity of genetic drift in individual species. This resource encompasses data for 1506 eukaryotic species, including 1413 animals and 93 green plants, and is organized in three components. The first two components contain approximations of the effective population size, which serve as indicators of the extent of random genetic drift within each species. In the first component, we meticulously investigated public databases to assemble data on life history traits such as longevity, adult body length and body mass for a set of 979 species. The second component includes estimations of the ratio between the rate of non-synonymous substitutions and the rate of synonymous substitutions (d<i>N</i>/d<i>S</i>) in protein-coding sequences for 1324 species. This ratio provides an estimate of the efficiency of natural selection in purging deleterious substitutions. Additionally, we present polymorphism-derived <i>N</i> <sub>e</sub> estimates for 66 species. The third component encompasses various genomic and transcriptomic characteristics. With this component, we aim to facilitate comparative transcriptomics analyses across species, by providing easy-to-use processed data for more than 16 000 RNA-seq samples across 491 species. These data include intron-centered alternative splicing frequencies, gene expression levels and sequencing depth statistics for each species, obtained with a homogeneous analysis protocol. To enable cross-species comparisons, we provide orthology predictions for conserved single-copy genes based on BUSCO gene sets. To illustrate the possible uses of this database, we identify the most frequently used introns for each gene and we assess how the sequencing depth available for each species affects our power to identify major and minor splice variants.</p>\",\"PeriodicalId\":33994,\"journal\":{\"name\":\"NAR Genomics and Bioinformatics\",\"volume\":\"6 2\",\"pages\":\"lqae064\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2024-06-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11167491/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"NAR Genomics and Bioinformatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1093/nargab/lqae064\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/6/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"NAR Genomics and Bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/nargab/lqae064","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/6/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
GTDrift: a resource for exploring the interplay between genetic drift, genomic and transcriptomic characteristics in eukaryotes.
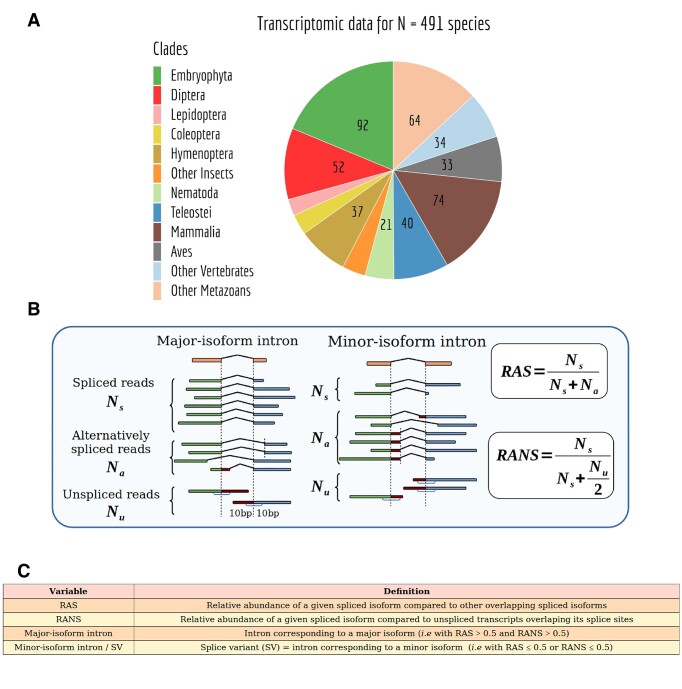
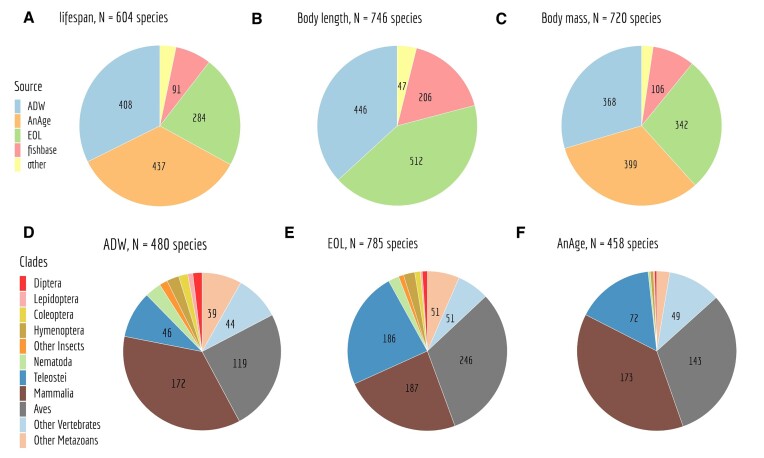
We present GTDrift, a comprehensive data resource that enables explorations of genomic and transcriptomic characteristics alongside proxies of the intensity of genetic drift in individual species. This resource encompasses data for 1506 eukaryotic species, including 1413 animals and 93 green plants, and is organized in three components. The first two components contain approximations of the effective population size, which serve as indicators of the extent of random genetic drift within each species. In the first component, we meticulously investigated public databases to assemble data on life history traits such as longevity, adult body length and body mass for a set of 979 species. The second component includes estimations of the ratio between the rate of non-synonymous substitutions and the rate of synonymous substitutions (dN/dS) in protein-coding sequences for 1324 species. This ratio provides an estimate of the efficiency of natural selection in purging deleterious substitutions. Additionally, we present polymorphism-derived Ne estimates for 66 species. The third component encompasses various genomic and transcriptomic characteristics. With this component, we aim to facilitate comparative transcriptomics analyses across species, by providing easy-to-use processed data for more than 16 000 RNA-seq samples across 491 species. These data include intron-centered alternative splicing frequencies, gene expression levels and sequencing depth statistics for each species, obtained with a homogeneous analysis protocol. To enable cross-species comparisons, we provide orthology predictions for conserved single-copy genes based on BUSCO gene sets. To illustrate the possible uses of this database, we identify the most frequently used introns for each gene and we assess how the sequencing depth available for each species affects our power to identify major and minor splice variants.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


