Colton D. Carlson, Jiarui Ma, Mohamad H. Al-Jabiri, Aran Insausti and Yunjie Xu
{"title":"1-苯基-2,2,2-三氟乙醇与两个水分子的构象适应和大振幅运动:旋转光谱和 Ab Initio 研究","authors":"Colton D. Carlson, Jiarui Ma, Mohamad H. Al-Jabiri, Aran Insausti and Yunjie Xu","doi":"10.1039/D4CP01516A","DOIUrl":null,"url":null,"abstract":"<p >The 1 : 2 adduct of 1-phenyl-2,2,2-trifluoroethanol (PhTFE), a chiral fluoroalcohol, with two water molecules (PhTFE⋯2H<small><sub>2</sub></small>O) was investigated <em>via</em> chirped pulse Fourier-transform microwave (CP-FTMW) spectroscopy and theoretical calculations. A systematic search of the PhTFE⋯2H<small><sub>2</sub></small>O conformational landscape identified 38 stable minima at the B3LYP-D3BJ/def2-TZVPPD level of theory, 27 of which are within an energy window of 10 kJ mol<small><sup>−1</sup></small> after applying zero-point energy corrections. Rotational spectra of a single PhTFE⋯2H<small><sub>2</sub></small>O conformer along with eight deuterated and three oxygen-18 isotopologues were assigned. Interestingly, the observed PhTFE⋯2H<small><sub>2</sub></small>O conformer contains PhTFE <strong>II</strong>, the second most stable monomer conformer, and the most stable PhTFE <strong>I</strong> dihydrate is <em>ca.</em> 4 kJ mol<small><sup>−1</sup></small> higher in energy. In contrast, PhTFE <strong>I</strong>⋯H<small><sub>2</sub></small>O was identified experimentally and theoretically as the most stable 1 : 1 conformer. Furthermore, the observed dihydrate structure experiences large amplitude motions connecting three theoretical minima which differ only in which water oxygen lone pairs are involved in the hydrogen-bonds, <em>i.e.</em>, the free OH pointing directions. Additionally, the <em>ortho</em> and <em>para</em>-H<small><sub>2</sub></small>O tunnelling splittings were detected and attributed to the interchange water hydrogen atoms which interact with the aromatic part of PhTFE but not for the water interacting with PhTFE hydroxy group. Extensive theoretical modelling was carried out to gain insight into the associated large amplitude motions including tunnelling, supported by the experimental isotopic and tunnelling splitting data.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 26","pages":" 18067-18075"},"PeriodicalIF":2.9000,"publicationDate":"2024-06-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Conformational adaptation and large amplitude motions of 1-phenyl-2,2,2-trifluoroethanol with two water molecules: a rotational spectroscopic and ab initio investigation†\",\"authors\":\"Colton D. Carlson, Jiarui Ma, Mohamad H. Al-Jabiri, Aran Insausti and Yunjie Xu\",\"doi\":\"10.1039/D4CP01516A\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The 1 : 2 adduct of 1-phenyl-2,2,2-trifluoroethanol (PhTFE), a chiral fluoroalcohol, with two water molecules (PhTFE⋯2H<small><sub>2</sub></small>O) was investigated <em>via</em> chirped pulse Fourier-transform microwave (CP-FTMW) spectroscopy and theoretical calculations. A systematic search of the PhTFE⋯2H<small><sub>2</sub></small>O conformational landscape identified 38 stable minima at the B3LYP-D3BJ/def2-TZVPPD level of theory, 27 of which are within an energy window of 10 kJ mol<small><sup>−1</sup></small> after applying zero-point energy corrections. Rotational spectra of a single PhTFE⋯2H<small><sub>2</sub></small>O conformer along with eight deuterated and three oxygen-18 isotopologues were assigned. Interestingly, the observed PhTFE⋯2H<small><sub>2</sub></small>O conformer contains PhTFE <strong>II</strong>, the second most stable monomer conformer, and the most stable PhTFE <strong>I</strong> dihydrate is <em>ca.</em> 4 kJ mol<small><sup>−1</sup></small> higher in energy. In contrast, PhTFE <strong>I</strong>⋯H<small><sub>2</sub></small>O was identified experimentally and theoretically as the most stable 1 : 1 conformer. Furthermore, the observed dihydrate structure experiences large amplitude motions connecting three theoretical minima which differ only in which water oxygen lone pairs are involved in the hydrogen-bonds, <em>i.e.</em>, the free OH pointing directions. Additionally, the <em>ortho</em> and <em>para</em>-H<small><sub>2</sub></small>O tunnelling splittings were detected and attributed to the interchange water hydrogen atoms which interact with the aromatic part of PhTFE but not for the water interacting with PhTFE hydroxy group. Extensive theoretical modelling was carried out to gain insight into the associated large amplitude motions including tunnelling, supported by the experimental isotopic and tunnelling splitting data.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 26\",\"pages\":\" 18067-18075\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2024-06-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2024/cp/d4cp01516a\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/cp/d4cp01516a","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Conformational adaptation and large amplitude motions of 1-phenyl-2,2,2-trifluoroethanol with two water molecules: a rotational spectroscopic and ab initio investigation†
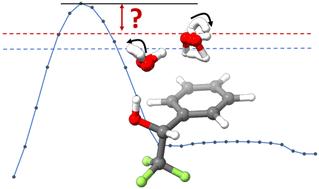
The 1 : 2 adduct of 1-phenyl-2,2,2-trifluoroethanol (PhTFE), a chiral fluoroalcohol, with two water molecules (PhTFE⋯2H2O) was investigated via chirped pulse Fourier-transform microwave (CP-FTMW) spectroscopy and theoretical calculations. A systematic search of the PhTFE⋯2H2O conformational landscape identified 38 stable minima at the B3LYP-D3BJ/def2-TZVPPD level of theory, 27 of which are within an energy window of 10 kJ mol−1 after applying zero-point energy corrections. Rotational spectra of a single PhTFE⋯2H2O conformer along with eight deuterated and three oxygen-18 isotopologues were assigned. Interestingly, the observed PhTFE⋯2H2O conformer contains PhTFE II, the second most stable monomer conformer, and the most stable PhTFE I dihydrate is ca. 4 kJ mol−1 higher in energy. In contrast, PhTFE I⋯H2O was identified experimentally and theoretically as the most stable 1 : 1 conformer. Furthermore, the observed dihydrate structure experiences large amplitude motions connecting three theoretical minima which differ only in which water oxygen lone pairs are involved in the hydrogen-bonds, i.e., the free OH pointing directions. Additionally, the ortho and para-H2O tunnelling splittings were detected and attributed to the interchange water hydrogen atoms which interact with the aromatic part of PhTFE but not for the water interacting with PhTFE hydroxy group. Extensive theoretical modelling was carried out to gain insight into the associated large amplitude motions including tunnelling, supported by the experimental isotopic and tunnelling splitting data.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


