Bianca R. Grosz, Jevin M. Parmar, Melina Ellis, Samantha Bryen, Cas Simons, Andre L. M. Reis, Igor Stevanovski, Ira W. Deveson, Garth Nicholson, Nigel Laing, Mathew Wallis, Gianina Ravenscroft, Kishore R. Kumar, Steve Vucic, Marina L. Kennerson
{"title":"MME 的一个深内含子变体通过异常剪接导致常染色体隐性夏科-玛丽-牙神经病。","authors":"Bianca R. Grosz, Jevin M. Parmar, Melina Ellis, Samantha Bryen, Cas Simons, Andre L. M. Reis, Igor Stevanovski, Ira W. Deveson, Garth Nicholson, Nigel Laing, Mathew Wallis, Gianina Ravenscroft, Kishore R. Kumar, Steve Vucic, Marina L. Kennerson","doi":"10.1111/jns.12637","DOIUrl":null,"url":null,"abstract":"<div>\n \n \n <section>\n \n <h3> Background</h3>\n \n <p>Loss-of-function variants in <i>MME</i> (membrane metalloendopeptidase) are a known cause of recessive Charcot–Marie–Tooth Neuropathy (CMT). A deep intronic variant, <i>MME</i> c.1188+428A>G (NM_000902.5), was identified through whole genome sequencing (WGS) of two Australian families with recessive inheritance of axonal CMT using the seqr platform. <i>MME</i> c.1188+428A>G was detected in a homozygous state in Family 1, and in a compound heterozygous state with a known pathogenic <i>MME</i> variant (c.467del; p.Pro156Leufs*14) in Family 2.</p>\n </section>\n \n <section>\n \n <h3> Aims</h3>\n \n <p>We aimed to determine the pathogenicity of the <i>MME</i> c.1188+428A>G variant through segregation and splicing analysis.</p>\n </section>\n \n <section>\n \n <h3> Methods</h3>\n \n <p>The splicing impact of the deep intronic <i>MME</i> variant c.1188+428A>G was assessed using an in vitro exon-trapping assay.</p>\n </section>\n \n <section>\n \n <h3> Results</h3>\n \n <p>The exon-trapping assay demonstrated that the <i>MME</i> c.1188+428A>G variant created a novel splice donor site resulting in the inclusion of an 83 bp pseudoexon between <i>MME</i> exons 12 and 13. The incorporation of the pseudoexon into <i>MME</i> transcript is predicted to lead to a coding frameshift and premature termination codon (PTC) in <i>MME</i> exon 14 (p.Ala397ProfsTer47). This PTC is likely to result in nonsense mediated decay (NMD) of <i>MME</i> transcript leading to a pathogenic loss-of-function.</p>\n </section>\n \n <section>\n \n <h3> Interpretation</h3>\n \n <p>To our knowledge, this is the first report of a pathogenic deep intronic <i>MME</i> variant causing CMT. This is of significance as deep intronic variants are missed using whole exome sequencing screening methods. Individuals with CMT should be reassessed for deep intronic variants, with splicing impacts being considered in relation to the potential pathogenicity of variants.</p>\n </section>\n </div>","PeriodicalId":17451,"journal":{"name":"Journal of the Peripheral Nervous System","volume":"29 2","pages":"262-274"},"PeriodicalIF":3.9000,"publicationDate":"2024-06-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/jns.12637","citationCount":"0","resultStr":"{\"title\":\"A deep intronic variant in MME causes autosomal recessive Charcot–Marie–Tooth neuropathy through aberrant splicing\",\"authors\":\"Bianca R. Grosz, Jevin M. Parmar, Melina Ellis, Samantha Bryen, Cas Simons, Andre L. M. Reis, Igor Stevanovski, Ira W. Deveson, Garth Nicholson, Nigel Laing, Mathew Wallis, Gianina Ravenscroft, Kishore R. Kumar, Steve Vucic, Marina L. Kennerson\",\"doi\":\"10.1111/jns.12637\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n \\n <section>\\n \\n <h3> Background</h3>\\n \\n <p>Loss-of-function variants in <i>MME</i> (membrane metalloendopeptidase) are a known cause of recessive Charcot–Marie–Tooth Neuropathy (CMT). A deep intronic variant, <i>MME</i> c.1188+428A>G (NM_000902.5), was identified through whole genome sequencing (WGS) of two Australian families with recessive inheritance of axonal CMT using the seqr platform. <i>MME</i> c.1188+428A>G was detected in a homozygous state in Family 1, and in a compound heterozygous state with a known pathogenic <i>MME</i> variant (c.467del; p.Pro156Leufs*14) in Family 2.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Aims</h3>\\n \\n <p>We aimed to determine the pathogenicity of the <i>MME</i> c.1188+428A>G variant through segregation and splicing analysis.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Methods</h3>\\n \\n <p>The splicing impact of the deep intronic <i>MME</i> variant c.1188+428A>G was assessed using an in vitro exon-trapping assay.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Results</h3>\\n \\n <p>The exon-trapping assay demonstrated that the <i>MME</i> c.1188+428A>G variant created a novel splice donor site resulting in the inclusion of an 83 bp pseudoexon between <i>MME</i> exons 12 and 13. The incorporation of the pseudoexon into <i>MME</i> transcript is predicted to lead to a coding frameshift and premature termination codon (PTC) in <i>MME</i> exon 14 (p.Ala397ProfsTer47). This PTC is likely to result in nonsense mediated decay (NMD) of <i>MME</i> transcript leading to a pathogenic loss-of-function.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Interpretation</h3>\\n \\n <p>To our knowledge, this is the first report of a pathogenic deep intronic <i>MME</i> variant causing CMT. This is of significance as deep intronic variants are missed using whole exome sequencing screening methods. Individuals with CMT should be reassessed for deep intronic variants, with splicing impacts being considered in relation to the potential pathogenicity of variants.</p>\\n </section>\\n </div>\",\"PeriodicalId\":17451,\"journal\":{\"name\":\"Journal of the Peripheral Nervous System\",\"volume\":\"29 2\",\"pages\":\"262-274\"},\"PeriodicalIF\":3.9000,\"publicationDate\":\"2024-06-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1111/jns.12637\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of the Peripheral Nervous System\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/jns.12637\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of the Peripheral Nervous System","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/jns.12637","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
A deep intronic variant in MME causes autosomal recessive Charcot–Marie–Tooth neuropathy through aberrant splicing
Background
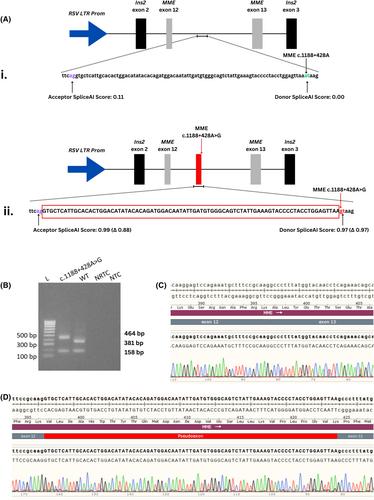
Loss-of-function variants in MME (membrane metalloendopeptidase) are a known cause of recessive Charcot–Marie–Tooth Neuropathy (CMT). A deep intronic variant, MME c.1188+428A>G (NM_000902.5), was identified through whole genome sequencing (WGS) of two Australian families with recessive inheritance of axonal CMT using the seqr platform. MME c.1188+428A>G was detected in a homozygous state in Family 1, and in a compound heterozygous state with a known pathogenic MME variant (c.467del; p.Pro156Leufs*14) in Family 2.
Aims
We aimed to determine the pathogenicity of the MME c.1188+428A>G variant through segregation and splicing analysis.
Methods
The splicing impact of the deep intronic MME variant c.1188+428A>G was assessed using an in vitro exon-trapping assay.
Results
The exon-trapping assay demonstrated that the MME c.1188+428A>G variant created a novel splice donor site resulting in the inclusion of an 83 bp pseudoexon between MME exons 12 and 13. The incorporation of the pseudoexon into MME transcript is predicted to lead to a coding frameshift and premature termination codon (PTC) in MME exon 14 (p.Ala397ProfsTer47). This PTC is likely to result in nonsense mediated decay (NMD) of MME transcript leading to a pathogenic loss-of-function.
Interpretation
To our knowledge, this is the first report of a pathogenic deep intronic MME variant causing CMT. This is of significance as deep intronic variants are missed using whole exome sequencing screening methods. Individuals with CMT should be reassessed for deep intronic variants, with splicing impacts being considered in relation to the potential pathogenicity of variants.
期刊介绍:
The Journal of the Peripheral Nervous System is the official journal of the Peripheral Nerve Society. Founded in 1996, it is the scientific journal of choice for clinicians, clinical scientists and basic neuroscientists interested in all aspects of biology and clinical research of peripheral nervous system disorders.
The Journal of the Peripheral Nervous System is a peer-reviewed journal that publishes high quality articles on cell and molecular biology, genomics, neuropathic pain, clinical research, trials, and unique case reports on inherited and acquired peripheral neuropathies.
Original articles are organized according to the topic in one of four specific areas: Mechanisms of Disease, Genetics, Clinical Research, and Clinical Trials.
The journal also publishes regular review papers on hot topics and Special Issues on basic, clinical, or assembled research in the field of peripheral nervous system disorders. Authors interested in contributing a review-type article or a Special Issue should contact the Editorial Office to discuss the scope of the proposed article with the Editor-in-Chief.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


