Aurore E. F. Denjean, Jordan Rio, Ilaria Ciofini, Marie-Eve L. Perrin, Pierre-Adrien Payard
{"title":"溶液中的计算能垒与实验能垒:密度泛函近似类型的影响。","authors":"Aurore E. F. Denjean, Jordan Rio, Ilaria Ciofini, Marie-Eve L. Perrin, Pierre-Adrien Payard","doi":"10.1002/jcc.27436","DOIUrl":null,"url":null,"abstract":"<p>Mechanistic investigations at the density functional theory level of organic and organometallic reactions in solution are now broadly accessible and routinely implemented to complement experimental investigations. The selection of an appropriate functional among the plethora of developed ones is the first challenge on the way to reliable energy barrier calculations. To provide guidelines for the choice of an initial and reliable computational level, the performances of commonly used non-empirical (PBE, PBE0, PBE0-DH) and empirical density functionals (BLYP, B3LYP, B2PLYP) were evaluated relative to experimental activation enthalpies. Most reactivity databases to assess density functional performances are primarily based on high level calculations, here a set of experimental activation enthalpies of organic and organometallic reactions performed in solution were selected from the literature. As a general trend, the non-empirical functionals outperform the empirical ones. The most accurate energy barriers are obtained with hybrid PBE0 and double-hybrid PBE0-DH density functionals, both providing similar performance. Regardless of the functional under consideration, the addition of the GD3-BJ empirical dispersion correction does not enhance the accuracy of computed energy barriers.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 27","pages":"2284-2293"},"PeriodicalIF":3.4000,"publicationDate":"2024-06-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Computed versus experimental energy barriers in solution: Influence of the type of the density functional approximation\",\"authors\":\"Aurore E. F. Denjean, Jordan Rio, Ilaria Ciofini, Marie-Eve L. Perrin, Pierre-Adrien Payard\",\"doi\":\"10.1002/jcc.27436\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Mechanistic investigations at the density functional theory level of organic and organometallic reactions in solution are now broadly accessible and routinely implemented to complement experimental investigations. The selection of an appropriate functional among the plethora of developed ones is the first challenge on the way to reliable energy barrier calculations. To provide guidelines for the choice of an initial and reliable computational level, the performances of commonly used non-empirical (PBE, PBE0, PBE0-DH) and empirical density functionals (BLYP, B3LYP, B2PLYP) were evaluated relative to experimental activation enthalpies. Most reactivity databases to assess density functional performances are primarily based on high level calculations, here a set of experimental activation enthalpies of organic and organometallic reactions performed in solution were selected from the literature. As a general trend, the non-empirical functionals outperform the empirical ones. The most accurate energy barriers are obtained with hybrid PBE0 and double-hybrid PBE0-DH density functionals, both providing similar performance. Regardless of the functional under consideration, the addition of the GD3-BJ empirical dispersion correction does not enhance the accuracy of computed energy barriers.</p>\",\"PeriodicalId\":188,\"journal\":{\"name\":\"Journal of Computational Chemistry\",\"volume\":\"45 27\",\"pages\":\"2284-2293\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2024-06-07\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computational Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27436\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27436","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
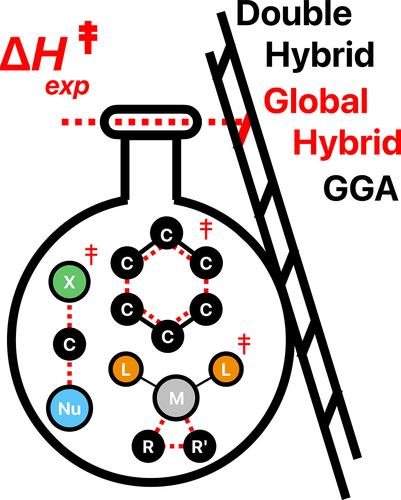
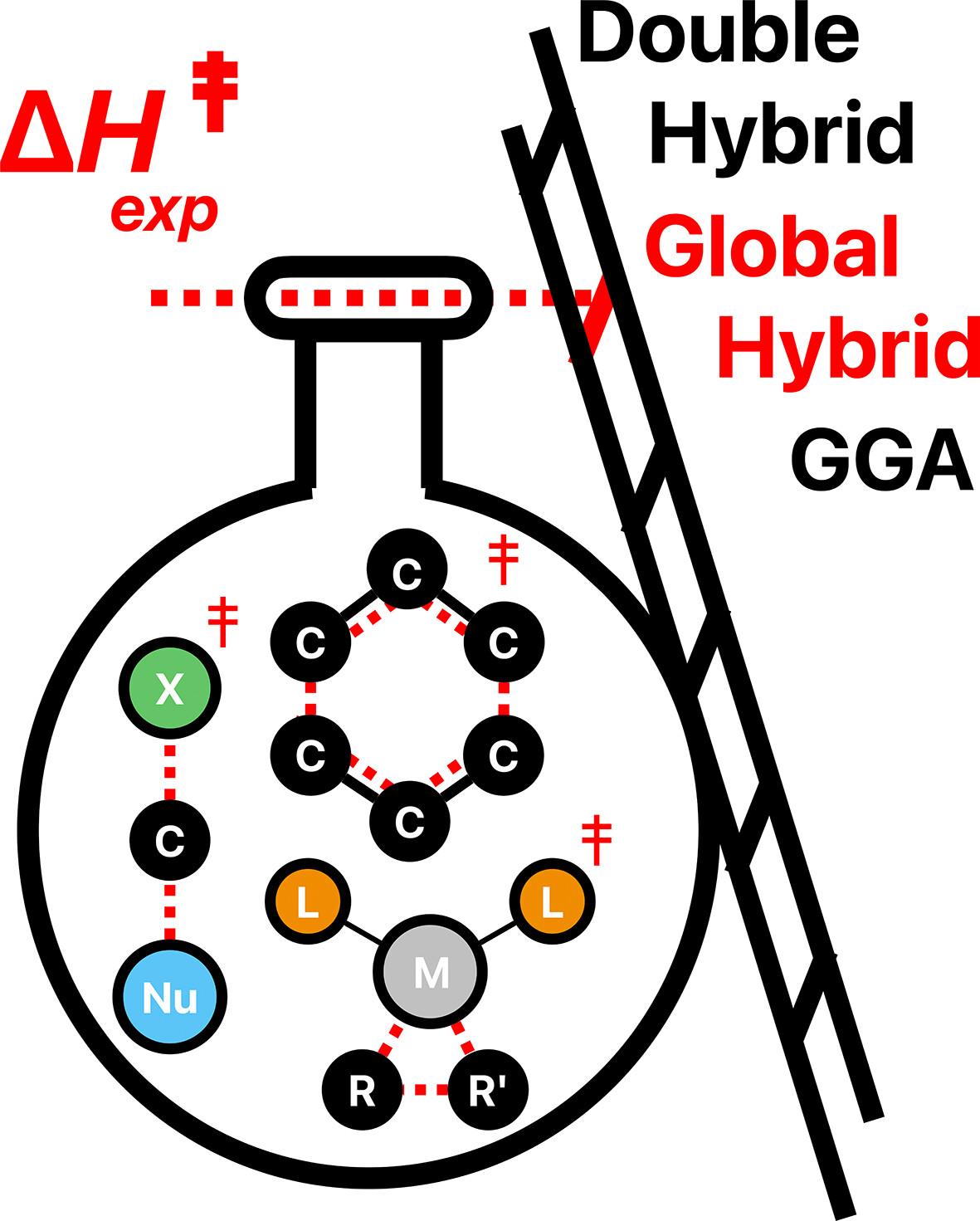
Computed versus experimental energy barriers in solution: Influence of the type of the density functional approximation
Mechanistic investigations at the density functional theory level of organic and organometallic reactions in solution are now broadly accessible and routinely implemented to complement experimental investigations. The selection of an appropriate functional among the plethora of developed ones is the first challenge on the way to reliable energy barrier calculations. To provide guidelines for the choice of an initial and reliable computational level, the performances of commonly used non-empirical (PBE, PBE0, PBE0-DH) and empirical density functionals (BLYP, B3LYP, B2PLYP) were evaluated relative to experimental activation enthalpies. Most reactivity databases to assess density functional performances are primarily based on high level calculations, here a set of experimental activation enthalpies of organic and organometallic reactions performed in solution were selected from the literature. As a general trend, the non-empirical functionals outperform the empirical ones. The most accurate energy barriers are obtained with hybrid PBE0 and double-hybrid PBE0-DH density functionals, both providing similar performance. Regardless of the functional under consideration, the addition of the GD3-BJ empirical dispersion correction does not enhance the accuracy of computed energy barriers.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


