Cailum M. K. Stienstra, Liam Hebert, Patrick Thomas, Alexander Haack, Jason Guo and W. Scott Hopkins*,
{"title":"Graphormer-IR:图形变换器利用高度专业化的注意力预测实验红外光谱。","authors":"Cailum M. K. Stienstra, Liam Hebert, Patrick Thomas, Alexander Haack, Jason Guo and W. Scott Hopkins*, ","doi":"10.1021/acs.jcim.4c00378","DOIUrl":null,"url":null,"abstract":"<p >Infrared (IR) spectroscopy is an important analytical tool in various chemical and forensic domains and a great deal of effort has gone into developing <i>in silico</i> methods for predicting experimental spectra. A key challenge in this regard is generating highly accurate spectra quickly to enable real-time feedback between computation and experiment. Here, we employ Graphormer, a graph neural network (GNN) transformer, to predict IR spectra using only simplified molecular-input line-entry system (SMILES) strings. Our data set includes 53,528 high-quality spectra, measured in five different experimental media (i.e., phases), for molecules containing the elements H, C, N, O, F, Si, S, P, Cl, Br, and I. When using only atomic numbers for node encodings, Graphormer-IR achieved a mean test spectral information similarity (<i>SIS</i><sub>μ</sub>) value of 0.8449 ± 0.0012 (<i>n</i> = 5), which surpasses that the current state-of-the-art model Chemprop-IR (<i>SIS</i><sub>μ</sub> = 0.8409 ± 0.0014, <i>n</i> = 5) with only 36% of the encoded information. Augmenting node embeddings with additional node-level descriptors in learned embeddings generated through a multilayer perceptron improves scores to <i>SIS</i><sub>μ</sub> = 0.8523 ± 0.0006, a total improvement of 19.7σ (<i>t</i> = 19). These improved scores show how Graphormer-IR excels in capturing long-range interactions like hydrogen bonding, anharmonic peak positions in experimental spectra, and stretching frequencies of uncommon functional groups. Scaling our architecture to 210 attention heads demonstrates specialist-like behavior for distinct IR frequencies that improves model performance. Our model utilizes novel architectures, including a global node for phase encoding, learned node feature embeddings, and a one-dimensional (1D) smoothing convolutional neural network (CNN). Graphormer-IR’s innovations underscore its value over traditional message-passing neural networks (MPNNs) due to its expressive embeddings and ability to capture long-range intramolecular relationships.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":null,"pages":null},"PeriodicalIF":5.6000,"publicationDate":"2024-06-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Graphormer-IR: Graph Transformers Predict Experimental IR Spectra Using Highly Specialized Attention\",\"authors\":\"Cailum M. K. Stienstra, Liam Hebert, Patrick Thomas, Alexander Haack, Jason Guo and W. Scott Hopkins*, \",\"doi\":\"10.1021/acs.jcim.4c00378\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Infrared (IR) spectroscopy is an important analytical tool in various chemical and forensic domains and a great deal of effort has gone into developing <i>in silico</i> methods for predicting experimental spectra. A key challenge in this regard is generating highly accurate spectra quickly to enable real-time feedback between computation and experiment. Here, we employ Graphormer, a graph neural network (GNN) transformer, to predict IR spectra using only simplified molecular-input line-entry system (SMILES) strings. Our data set includes 53,528 high-quality spectra, measured in five different experimental media (i.e., phases), for molecules containing the elements H, C, N, O, F, Si, S, P, Cl, Br, and I. When using only atomic numbers for node encodings, Graphormer-IR achieved a mean test spectral information similarity (<i>SIS</i><sub>μ</sub>) value of 0.8449 ± 0.0012 (<i>n</i> = 5), which surpasses that the current state-of-the-art model Chemprop-IR (<i>SIS</i><sub>μ</sub> = 0.8409 ± 0.0014, <i>n</i> = 5) with only 36% of the encoded information. Augmenting node embeddings with additional node-level descriptors in learned embeddings generated through a multilayer perceptron improves scores to <i>SIS</i><sub>μ</sub> = 0.8523 ± 0.0006, a total improvement of 19.7σ (<i>t</i> = 19). These improved scores show how Graphormer-IR excels in capturing long-range interactions like hydrogen bonding, anharmonic peak positions in experimental spectra, and stretching frequencies of uncommon functional groups. Scaling our architecture to 210 attention heads demonstrates specialist-like behavior for distinct IR frequencies that improves model performance. Our model utilizes novel architectures, including a global node for phase encoding, learned node feature embeddings, and a one-dimensional (1D) smoothing convolutional neural network (CNN). Graphormer-IR’s innovations underscore its value over traditional message-passing neural networks (MPNNs) due to its expressive embeddings and ability to capture long-range intramolecular relationships.</p>\",\"PeriodicalId\":44,\"journal\":{\"name\":\"Journal of Chemical Information and Modeling \",\"volume\":null,\"pages\":null},\"PeriodicalIF\":5.6000,\"publicationDate\":\"2024-06-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Information and Modeling \",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jcim.4c00378\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jcim.4c00378","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
Graphormer-IR: Graph Transformers Predict Experimental IR Spectra Using Highly Specialized Attention
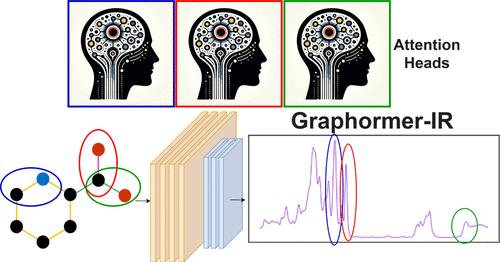
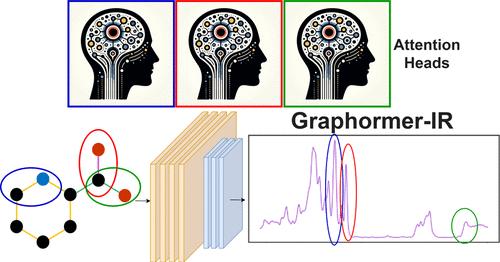
Infrared (IR) spectroscopy is an important analytical tool in various chemical and forensic domains and a great deal of effort has gone into developing in silico methods for predicting experimental spectra. A key challenge in this regard is generating highly accurate spectra quickly to enable real-time feedback between computation and experiment. Here, we employ Graphormer, a graph neural network (GNN) transformer, to predict IR spectra using only simplified molecular-input line-entry system (SMILES) strings. Our data set includes 53,528 high-quality spectra, measured in five different experimental media (i.e., phases), for molecules containing the elements H, C, N, O, F, Si, S, P, Cl, Br, and I. When using only atomic numbers for node encodings, Graphormer-IR achieved a mean test spectral information similarity (SISμ) value of 0.8449 ± 0.0012 (n = 5), which surpasses that the current state-of-the-art model Chemprop-IR (SISμ = 0.8409 ± 0.0014, n = 5) with only 36% of the encoded information. Augmenting node embeddings with additional node-level descriptors in learned embeddings generated through a multilayer perceptron improves scores to SISμ = 0.8523 ± 0.0006, a total improvement of 19.7σ (t = 19). These improved scores show how Graphormer-IR excels in capturing long-range interactions like hydrogen bonding, anharmonic peak positions in experimental spectra, and stretching frequencies of uncommon functional groups. Scaling our architecture to 210 attention heads demonstrates specialist-like behavior for distinct IR frequencies that improves model performance. Our model utilizes novel architectures, including a global node for phase encoding, learned node feature embeddings, and a one-dimensional (1D) smoothing convolutional neural network (CNN). Graphormer-IR’s innovations underscore its value over traditional message-passing neural networks (MPNNs) due to its expressive embeddings and ability to capture long-range intramolecular relationships.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


