Abhijit Dutta , Amit Kumar Pradhan , Paritosh Mondal
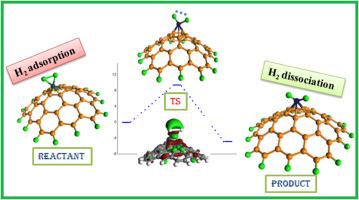
{"title":"活性碳支撑过渡金属原子对分子氢吸附和解离的催化作用:量子化学在可再生能源领域的应用进展","authors":"Abhijit Dutta , Amit Kumar Pradhan , Paritosh Mondal","doi":"10.1016/j.jmgm.2024.108804","DOIUrl":null,"url":null,"abstract":"<div><p>Density functional theory (DFT) investigation has been done to unravel the adsorption and dissociation nature of hydrogen molecule on 3<em>d</em>, 4<em>d</em> and 5<em>d</em> transition metal (M = Fe, Co, Ni, Cu, Ru, Rh, Pd, Ag, Os, Ir, Pt or Au) atom doped activated carbon (AC) surface. Transition metal doped AC are found to be active catalyst for storage of hydrogen and also gives the stability of <em>M</em> − H bonds formed after bond breakage of H<sub>2</sub> molecule. Transition metals are found to occupy the position on the five member ring rather than six member ring of the AC. Five member ring of the AC is seen to be more deformed than the six-member ring on metal doping. Higher values of LUMO-HOMO gap and vertical ionization potential and lower electron affinity signify the higher stability of hydrogen molecule adsorbed metal doped AC. Bond length and vibrational analysis of the adsorbed hydrogen molecule suggest the higher activation of hydrogen molecule on AC, where 4<em>d</em> and 5<em>d</em> metal doped ACs are found to be more efficient in comparison to 3<em>d</em> metal. Adsorbed hydrogen molecule on metal doped AC follows dissociation either via spill-over or via normal process. DFT evaluated rate constant and the transition states suggest that Ru, Rh, Os and Ir doped AC are found to be efficient in the dissociation of hydrogen molecule, while, Cu doped AC is seen to be worst in the same reaction. Deformed electron density, HOMO-LUMO isosurface, and density of states confirms the redistribution of electrons among H<sub>2</sub> and metal doped AC surface. ΔG<sub>H</sub> values of Hydrogen evolution reaction also signifies the greater catalytic activities of Ru and Os supported activated carbon towards HER.</p></div>","PeriodicalId":16361,"journal":{"name":"Journal of molecular graphics & modelling","volume":"131 ","pages":"Article 108804"},"PeriodicalIF":2.7000,"publicationDate":"2024-06-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Catalytic battle of activated carbon supported transition metal atom towards adsorption and dissociation of molecular hydrogen: Progress towards quantum chemical application on renewable energy resource\",\"authors\":\"Abhijit Dutta , Amit Kumar Pradhan , Paritosh Mondal\",\"doi\":\"10.1016/j.jmgm.2024.108804\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Density functional theory (DFT) investigation has been done to unravel the adsorption and dissociation nature of hydrogen molecule on 3<em>d</em>, 4<em>d</em> and 5<em>d</em> transition metal (M = Fe, Co, Ni, Cu, Ru, Rh, Pd, Ag, Os, Ir, Pt or Au) atom doped activated carbon (AC) surface. Transition metal doped AC are found to be active catalyst for storage of hydrogen and also gives the stability of <em>M</em> − H bonds formed after bond breakage of H<sub>2</sub> molecule. Transition metals are found to occupy the position on the five member ring rather than six member ring of the AC. Five member ring of the AC is seen to be more deformed than the six-member ring on metal doping. Higher values of LUMO-HOMO gap and vertical ionization potential and lower electron affinity signify the higher stability of hydrogen molecule adsorbed metal doped AC. Bond length and vibrational analysis of the adsorbed hydrogen molecule suggest the higher activation of hydrogen molecule on AC, where 4<em>d</em> and 5<em>d</em> metal doped ACs are found to be more efficient in comparison to 3<em>d</em> metal. Adsorbed hydrogen molecule on metal doped AC follows dissociation either via spill-over or via normal process. DFT evaluated rate constant and the transition states suggest that Ru, Rh, Os and Ir doped AC are found to be efficient in the dissociation of hydrogen molecule, while, Cu doped AC is seen to be worst in the same reaction. Deformed electron density, HOMO-LUMO isosurface, and density of states confirms the redistribution of electrons among H<sub>2</sub> and metal doped AC surface. ΔG<sub>H</sub> values of Hydrogen evolution reaction also signifies the greater catalytic activities of Ru and Os supported activated carbon towards HER.</p></div>\",\"PeriodicalId\":16361,\"journal\":{\"name\":\"Journal of molecular graphics & modelling\",\"volume\":\"131 \",\"pages\":\"Article 108804\"},\"PeriodicalIF\":2.7000,\"publicationDate\":\"2024-06-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of molecular graphics & modelling\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S1093326324001049\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular graphics & modelling","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1093326324001049","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
摘要
密度泛函理论(DFT)研究揭示了氢分子在 3d、4d 和 5d 过渡金属(M = Fe、Co、Ni、Cu、Ru、Rh、Pd、Ag、Os、Ir、Pt 或 Au)原子掺杂的活性碳(AC)表面的吸附和解离性质。研究发现,掺杂过渡金属的活性炭是储存氢气的活性催化剂,还能使氢分子断键后形成的 M - H 键保持稳定。过渡金属被发现位于 AC 的五元环上,而不是六元环上。在掺杂金属时,AC 的五元环比六元环变形更大。较高的 LUMO-HOMO 间隙和垂直电离电位值以及较低的电子亲和力表明氢分子吸附在掺杂金属的 AC 上具有更高的稳定性。吸附氢分子的键长和振动分析表明,氢分子在 AC 上的活化程度较高,其中掺杂 4d 和 5d 金属的 AC 比掺杂 3d 金属的 AC 更有效。掺杂金属的 AC 上吸附的氢分子通过溢出或正常过程解离。DFT 评估的速率常数和过渡态表明,掺杂 Ru、Rh、Os 和 Ir 的 AC 在氢分子解离过程中效率较高,而掺杂 Cu 的 AC 在相同反应中效率最差。变形电子密度、HOMO-LUMO 等位面和态密度证实了电子在氢气和掺杂金属的 AC 表面之间的重新分布。氢气进化反应的 ΔGH 值也表明 Ru 和 Os 支持的活性炭对氢气进化反应具有更强的催化活性。
Catalytic battle of activated carbon supported transition metal atom towards adsorption and dissociation of molecular hydrogen: Progress towards quantum chemical application on renewable energy resource
Density functional theory (DFT) investigation has been done to unravel the adsorption and dissociation nature of hydrogen molecule on 3d, 4d and 5d transition metal (M = Fe, Co, Ni, Cu, Ru, Rh, Pd, Ag, Os, Ir, Pt or Au) atom doped activated carbon (AC) surface. Transition metal doped AC are found to be active catalyst for storage of hydrogen and also gives the stability of M − H bonds formed after bond breakage of H2 molecule. Transition metals are found to occupy the position on the five member ring rather than six member ring of the AC. Five member ring of the AC is seen to be more deformed than the six-member ring on metal doping. Higher values of LUMO-HOMO gap and vertical ionization potential and lower electron affinity signify the higher stability of hydrogen molecule adsorbed metal doped AC. Bond length and vibrational analysis of the adsorbed hydrogen molecule suggest the higher activation of hydrogen molecule on AC, where 4d and 5d metal doped ACs are found to be more efficient in comparison to 3d metal. Adsorbed hydrogen molecule on metal doped AC follows dissociation either via spill-over or via normal process. DFT evaluated rate constant and the transition states suggest that Ru, Rh, Os and Ir doped AC are found to be efficient in the dissociation of hydrogen molecule, while, Cu doped AC is seen to be worst in the same reaction. Deformed electron density, HOMO-LUMO isosurface, and density of states confirms the redistribution of electrons among H2 and metal doped AC surface. ΔGH values of Hydrogen evolution reaction also signifies the greater catalytic activities of Ru and Os supported activated carbon towards HER.
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


