{"title":"用于药物重新定位的知识图谱卷积网络与启发式搜索","authors":"Xiang Du, Xinliang Sun and Min Li*, ","doi":"10.1021/acs.jcim.4c00737","DOIUrl":null,"url":null,"abstract":"<p >Drug repositioning is a strategy of repurposing approved drugs for treating new indications, which can accelerate the drug discovery process, reduce development costs, and lower the safety risk. The advancement of biotechnology has significantly accelerated the speed and scale of biological data generation, offering significant potential for drug repositioning through biomedical knowledge graphs that integrate diverse entities and relations from various biomedical sources. To fully learn the semantic information and topological structure information from the biological knowledge graph, we propose a knowledge graph convolutional network with a heuristic search, named KGCNH, which can effectively utilize the diversity of entities and relationships in biological knowledge graphs, as well as topological structure information, to predict the associations between drugs and diseases. Specifically, we design a relation-aware attention mechanism to compute the attention scores for each neighboring entity of a given entity under different relations. To address the challenge of randomness of the initial attention scores potentially impacting model performance and to expand the search scope of the model, we designed a heuristic search module based on Gumbel-Softmax, which uses attention scores as heuristic information and introduces randomness to assist the model in exploring more optimal embeddings of drugs and diseases. Following this module, we derive the relation weights, obtain the embeddings of drugs and diseases through neighborhood aggregation, and then predict drug–disease associations. Additionally, we employ feature-based augmented views to enhance model robustness and mitigate overfitting issues. We have implemented our method and conducted experiments on two data sets. The results demonstrate that KGCNH outperforms competing methods. In particular, case studies on lithium and quetiapine confirm that KGCNH can retrieve more actual drug–disease associations in the top prediction results.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":null,"pages":null},"PeriodicalIF":5.6000,"publicationDate":"2024-06-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Knowledge Graph Convolutional Network with Heuristic Search for Drug Repositioning\",\"authors\":\"Xiang Du, Xinliang Sun and Min Li*, \",\"doi\":\"10.1021/acs.jcim.4c00737\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Drug repositioning is a strategy of repurposing approved drugs for treating new indications, which can accelerate the drug discovery process, reduce development costs, and lower the safety risk. The advancement of biotechnology has significantly accelerated the speed and scale of biological data generation, offering significant potential for drug repositioning through biomedical knowledge graphs that integrate diverse entities and relations from various biomedical sources. To fully learn the semantic information and topological structure information from the biological knowledge graph, we propose a knowledge graph convolutional network with a heuristic search, named KGCNH, which can effectively utilize the diversity of entities and relationships in biological knowledge graphs, as well as topological structure information, to predict the associations between drugs and diseases. Specifically, we design a relation-aware attention mechanism to compute the attention scores for each neighboring entity of a given entity under different relations. To address the challenge of randomness of the initial attention scores potentially impacting model performance and to expand the search scope of the model, we designed a heuristic search module based on Gumbel-Softmax, which uses attention scores as heuristic information and introduces randomness to assist the model in exploring more optimal embeddings of drugs and diseases. Following this module, we derive the relation weights, obtain the embeddings of drugs and diseases through neighborhood aggregation, and then predict drug–disease associations. Additionally, we employ feature-based augmented views to enhance model robustness and mitigate overfitting issues. We have implemented our method and conducted experiments on two data sets. The results demonstrate that KGCNH outperforms competing methods. In particular, case studies on lithium and quetiapine confirm that KGCNH can retrieve more actual drug–disease associations in the top prediction results.</p>\",\"PeriodicalId\":44,\"journal\":{\"name\":\"Journal of Chemical Information and Modeling \",\"volume\":null,\"pages\":null},\"PeriodicalIF\":5.6000,\"publicationDate\":\"2024-06-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Information and Modeling \",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jcim.4c00737\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jcim.4c00737","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
Knowledge Graph Convolutional Network with Heuristic Search for Drug Repositioning
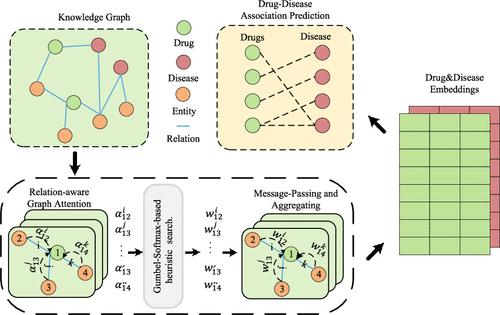
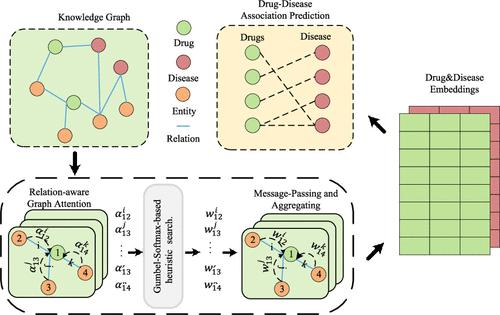
Drug repositioning is a strategy of repurposing approved drugs for treating new indications, which can accelerate the drug discovery process, reduce development costs, and lower the safety risk. The advancement of biotechnology has significantly accelerated the speed and scale of biological data generation, offering significant potential for drug repositioning through biomedical knowledge graphs that integrate diverse entities and relations from various biomedical sources. To fully learn the semantic information and topological structure information from the biological knowledge graph, we propose a knowledge graph convolutional network with a heuristic search, named KGCNH, which can effectively utilize the diversity of entities and relationships in biological knowledge graphs, as well as topological structure information, to predict the associations between drugs and diseases. Specifically, we design a relation-aware attention mechanism to compute the attention scores for each neighboring entity of a given entity under different relations. To address the challenge of randomness of the initial attention scores potentially impacting model performance and to expand the search scope of the model, we designed a heuristic search module based on Gumbel-Softmax, which uses attention scores as heuristic information and introduces randomness to assist the model in exploring more optimal embeddings of drugs and diseases. Following this module, we derive the relation weights, obtain the embeddings of drugs and diseases through neighborhood aggregation, and then predict drug–disease associations. Additionally, we employ feature-based augmented views to enhance model robustness and mitigate overfitting issues. We have implemented our method and conducted experiments on two data sets. The results demonstrate that KGCNH outperforms competing methods. In particular, case studies on lithium and quetiapine confirm that KGCNH can retrieve more actual drug–disease associations in the top prediction results.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


