Vivianne K. Ocampo-Restrepo, Sudarshan Vijay, G. T. Kasun Kalhara Gunasooriya and Jens K. Nørskov
{"title":"通过温度编程 O2 解吸模拟确定 IrO2 吸附位点的特征","authors":"Vivianne K. Ocampo-Restrepo, Sudarshan Vijay, G. T. Kasun Kalhara Gunasooriya and Jens K. Nørskov","doi":"10.1039/D4CP01213E","DOIUrl":null,"url":null,"abstract":"<p >This study presents simulations of temperature-programmed desorption (TPD) profiles using desorption energy data from density functional theory (DFT) calculations. We apply this method to investigate the desorption of oxygen (O<small><sub>2</sub></small>) from IrO<small><sub>2</sub></small>(110) to gain insight into the kinetics of oxygen coupling and desorption, important elementary steps in the oxygen evolution reaction (OER). Initially, we confirm the thermodynamically stable adsorption site for oxygen in the pristine IrO<small><sub>2</sub></small>(110) as Ir<small><sub>CUS</sub></small>, even with a high oxygen coverage. We successfully simulate TPD for O<small><sub>2</sub></small> desorption, achieving good agreement with experimental TPD data for different initial oxygen exposures when including more than one adsorption site. We identify a new adsorption site, related to the formation of steps on IrO<small><sub>2</sub></small>(110)(Ir<small><sub>CUS-step-0.5</sub></small>), that is essential for reproducing the experimental TPD. Our findings suggest that the observed TPD peaks are the result of different adsorption sites on the surface, rather than solely a lateral interactions effect. This work provides insight into the behavior of oxygen adsorption on IrO<small><sub>2</sub></small>, with implications for understanding surface reactivity and catalytic processes involving this material.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 24","pages":" 17396-17404"},"PeriodicalIF":2.9000,"publicationDate":"2024-06-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Characterization of adsorption sites on IrO2via temperature programmed O2 desorption simulations†\",\"authors\":\"Vivianne K. Ocampo-Restrepo, Sudarshan Vijay, G. T. Kasun Kalhara Gunasooriya and Jens K. Nørskov\",\"doi\":\"10.1039/D4CP01213E\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >This study presents simulations of temperature-programmed desorption (TPD) profiles using desorption energy data from density functional theory (DFT) calculations. We apply this method to investigate the desorption of oxygen (O<small><sub>2</sub></small>) from IrO<small><sub>2</sub></small>(110) to gain insight into the kinetics of oxygen coupling and desorption, important elementary steps in the oxygen evolution reaction (OER). Initially, we confirm the thermodynamically stable adsorption site for oxygen in the pristine IrO<small><sub>2</sub></small>(110) as Ir<small><sub>CUS</sub></small>, even with a high oxygen coverage. We successfully simulate TPD for O<small><sub>2</sub></small> desorption, achieving good agreement with experimental TPD data for different initial oxygen exposures when including more than one adsorption site. We identify a new adsorption site, related to the formation of steps on IrO<small><sub>2</sub></small>(110)(Ir<small><sub>CUS-step-0.5</sub></small>), that is essential for reproducing the experimental TPD. Our findings suggest that the observed TPD peaks are the result of different adsorption sites on the surface, rather than solely a lateral interactions effect. This work provides insight into the behavior of oxygen adsorption on IrO<small><sub>2</sub></small>, with implications for understanding surface reactivity and catalytic processes involving this material.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 24\",\"pages\":\" 17396-17404\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2024-06-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2024/cp/d4cp01213e\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/cp/d4cp01213e","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
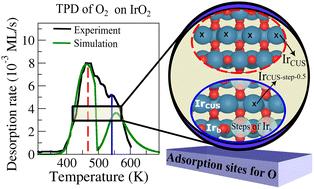
Characterization of adsorption sites on IrO2via temperature programmed O2 desorption simulations†
This study presents simulations of temperature-programmed desorption (TPD) profiles using desorption energy data from density functional theory (DFT) calculations. We apply this method to investigate the desorption of oxygen (O2) from IrO2(110) to gain insight into the kinetics of oxygen coupling and desorption, important elementary steps in the oxygen evolution reaction (OER). Initially, we confirm the thermodynamically stable adsorption site for oxygen in the pristine IrO2(110) as IrCUS, even with a high oxygen coverage. We successfully simulate TPD for O2 desorption, achieving good agreement with experimental TPD data for different initial oxygen exposures when including more than one adsorption site. We identify a new adsorption site, related to the formation of steps on IrO2(110)(IrCUS-step-0.5), that is essential for reproducing the experimental TPD. Our findings suggest that the observed TPD peaks are the result of different adsorption sites on the surface, rather than solely a lateral interactions effect. This work provides insight into the behavior of oxygen adsorption on IrO2, with implications for understanding surface reactivity and catalytic processes involving this material.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


