{"title":"灵活的调控网络建模改进了转录因子活性估计。","authors":"Chen Chen, Megha Padi","doi":"10.1038/s41540-024-00386-w","DOIUrl":null,"url":null,"abstract":"<p><p>Transcriptional regulation plays a crucial role in determining cell fate and disease, yet inferring the key regulators from gene expression data remains a significant challenge. Existing methods for estimating transcription factor (TF) activity often rely on static TF-gene interaction databases and cannot adapt to changes in regulatory mechanisms across different cell types and disease conditions. Here, we present a new algorithm - Transcriptional Inference using Gene Expression and Regulatory data (TIGER) - that overcomes these limitations by flexibly modeling activation and inhibition events, up-weighting essential edges, shrinking irrelevant edges towards zero through a sparse Bayesian prior, and simultaneously estimating both TF activity levels and changes in the underlying regulatory network. When applied to yeast and cancer TF knock-out datasets, TIGER outperforms comparable methods in terms of prediction accuracy. Moreover, our application of TIGER to tissue- and cell-type-specific RNA-seq data demonstrates its ability to uncover differences in regulatory mechanisms. Collectively, our findings highlight the utility of modeling context-specific regulation when inferring transcription factor activities.</p>","PeriodicalId":19345,"journal":{"name":"NPJ Systems Biology and Applications","volume":null,"pages":null},"PeriodicalIF":3.5000,"publicationDate":"2024-05-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11133322/pdf/","citationCount":"0","resultStr":"{\"title\":\"Flexible modeling of regulatory networks improves transcription factor activity estimation.\",\"authors\":\"Chen Chen, Megha Padi\",\"doi\":\"10.1038/s41540-024-00386-w\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Transcriptional regulation plays a crucial role in determining cell fate and disease, yet inferring the key regulators from gene expression data remains a significant challenge. Existing methods for estimating transcription factor (TF) activity often rely on static TF-gene interaction databases and cannot adapt to changes in regulatory mechanisms across different cell types and disease conditions. Here, we present a new algorithm - Transcriptional Inference using Gene Expression and Regulatory data (TIGER) - that overcomes these limitations by flexibly modeling activation and inhibition events, up-weighting essential edges, shrinking irrelevant edges towards zero through a sparse Bayesian prior, and simultaneously estimating both TF activity levels and changes in the underlying regulatory network. When applied to yeast and cancer TF knock-out datasets, TIGER outperforms comparable methods in terms of prediction accuracy. Moreover, our application of TIGER to tissue- and cell-type-specific RNA-seq data demonstrates its ability to uncover differences in regulatory mechanisms. Collectively, our findings highlight the utility of modeling context-specific regulation when inferring transcription factor activities.</p>\",\"PeriodicalId\":19345,\"journal\":{\"name\":\"NPJ Systems Biology and Applications\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":3.5000,\"publicationDate\":\"2024-05-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11133322/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"NPJ Systems Biology and Applications\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1038/s41540-024-00386-w\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"NPJ Systems Biology and Applications","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1038/s41540-024-00386-w","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
Flexible modeling of regulatory networks improves transcription factor activity estimation.
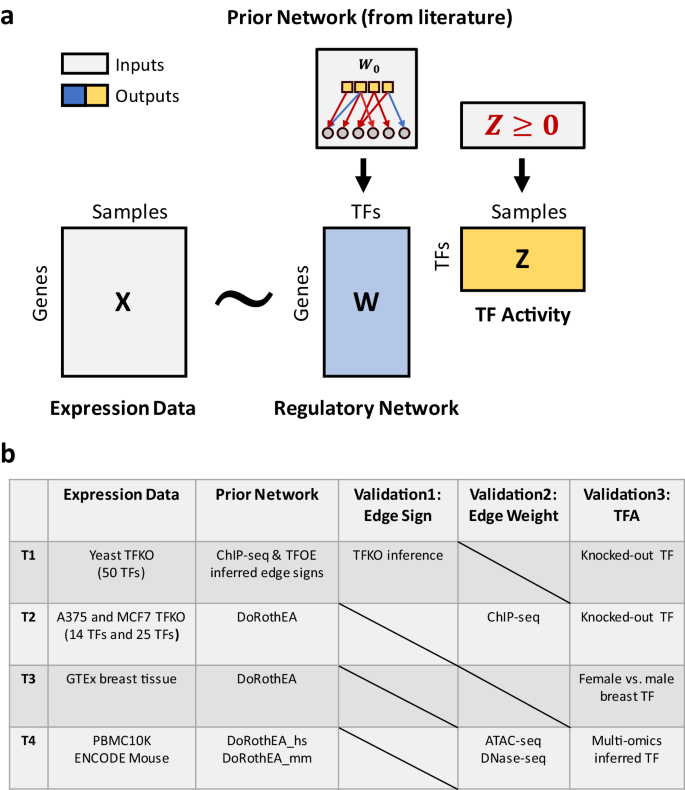
Transcriptional regulation plays a crucial role in determining cell fate and disease, yet inferring the key regulators from gene expression data remains a significant challenge. Existing methods for estimating transcription factor (TF) activity often rely on static TF-gene interaction databases and cannot adapt to changes in regulatory mechanisms across different cell types and disease conditions. Here, we present a new algorithm - Transcriptional Inference using Gene Expression and Regulatory data (TIGER) - that overcomes these limitations by flexibly modeling activation and inhibition events, up-weighting essential edges, shrinking irrelevant edges towards zero through a sparse Bayesian prior, and simultaneously estimating both TF activity levels and changes in the underlying regulatory network. When applied to yeast and cancer TF knock-out datasets, TIGER outperforms comparable methods in terms of prediction accuracy. Moreover, our application of TIGER to tissue- and cell-type-specific RNA-seq data demonstrates its ability to uncover differences in regulatory mechanisms. Collectively, our findings highlight the utility of modeling context-specific regulation when inferring transcription factor activities.
期刊介绍:
npj Systems Biology and Applications is an online Open Access journal dedicated to publishing the premier research that takes a systems-oriented approach. The journal aims to provide a forum for the presentation of articles that help define this nascent field, as well as those that apply the advances to wider fields. We encourage studies that integrate, or aid the integration of, data, analyses and insight from molecules to organisms and broader systems. Important areas of interest include not only fundamental biological systems and drug discovery, but also applications to health, medical practice and implementation, big data, biotechnology, food science, human behaviour, broader biological systems and industrial applications of systems biology.
We encourage all approaches, including network biology, application of control theory to biological systems, computational modelling and analysis, comprehensive and/or high-content measurements, theoretical, analytical and computational studies of system-level properties of biological systems and computational/software/data platforms enabling such studies.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


