APF2:一种用于药物基因组变异效应预测的改进型集合方法。
IF 2.9
3区 医学
Q2 GENETICS & HEREDITY
引用次数: 0
摘要
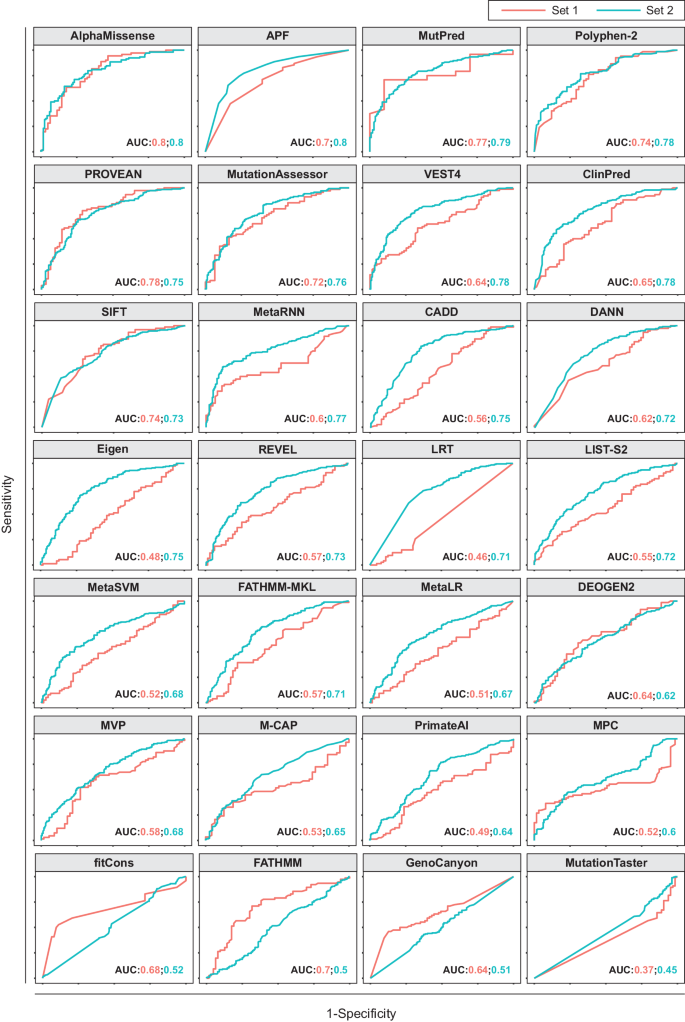
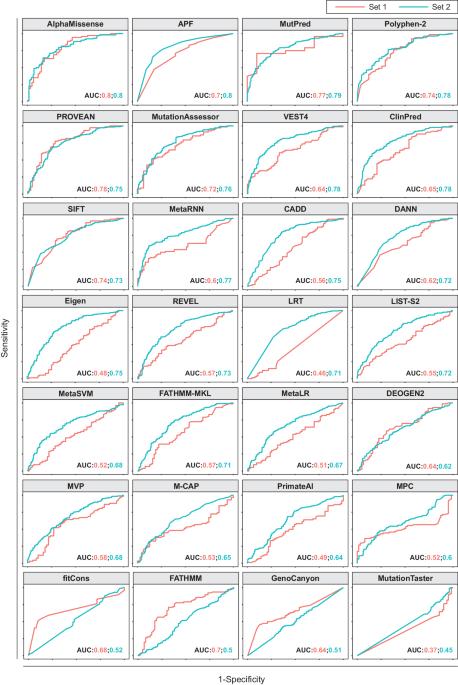
缺乏疗效或药物不良反应是药物治疗中的常见现象,会导致相当高的发病率和死亡率。据估计,在药物反应的变异中,有 20%-30% 源自编码药物靶点的基因或药物处置相关因素的变异。因此,利用这些药物基因组学信息来预先识别哪些患者可从剂量调整或替代药物中获益,是精准医疗的一个重要前沿领域。计算方法可用于预测意义不明的变异体的功能效应。然而,这些方法在药物基因组变异数据上的表现却乏善可陈。为了克服这一局限,我们之前开发了一种专门用于药物基因组变异预测的集合分类器,称为 APF。在此,我们希望利用最近在基于深度神经网络的蛋白质折叠预测方面取得的重要进展,进一步改进预测结果。在 530 个药物基因组错义变体上对 28 个变体效应预测器进行的基准测试表明,使用 AlphaMissense 进行的结构预测最具特异性,而 APF 则表现出最均衡的性能。随后,我们开发了一种新工具 APF2,通过优化针对药物基因组变异的最佳性能算法的算法参数,并将其预测结果汇总为统一的集合得分。重要的是,APF2 提供的定量变异效应估计值与实验结果有很好的相关性(R2 = 0.91,p = 0.003),并能预测药物基因组变异的功能影响,其准确性高于以前的方法,特别是对于具有可操作药物基因组指南的临床相关变异。此外,我们还在未用于模型训练或验证的、由 61 个药物基因中的 146 个变异组成的独立测试集上展示了更好的性能(92% 的准确率)。将 APF2 应用于来自 80 多万人的人群规模测序数据,发现了巨大的人种地理差异,对药物治疗具有重要影响。因此,我们认为 APF2 有潜力将遗传信息转化为药物遗传学建议,从而促进下一代测序数据在分层医疗中的应用。本文章由计算机程序翻译,如有差异,请以英文原文为准。
APF2: an improved ensemble method for pharmacogenomic variant effect prediction
Lack of efficacy or adverse drug response are common phenomena in pharmacological therapy causing considerable morbidity and mortality. It is estimated that 20–30% of this variability in drug response stems from variations in genes encoding drug targets or factors involved in drug disposition. Leveraging such pharmacogenomic information for the preemptive identification of patients who would benefit from dose adjustments or alternative medications thus constitutes an important frontier of precision medicine. Computational methods can be used to predict the functional effects of variant of unknown significance. However, their performance on pharmacogenomic variant data has been lackluster. To overcome this limitation, we previously developed an ensemble classifier, termed APF, specifically designed for pharmacogenomic variant prediction. Here, we aimed to further improve predictions by leveraging recent key advances in the prediction of protein folding based on deep neural networks. Benchmarking of 28 variant effect predictors on 530 pharmacogenetic missense variants revealed that structural predictions using AlphaMissense were most specific, whereas APF exhibited the most balanced performance. We then developed a new tool, APF2, by optimizing algorithm parametrization of the top performing algorithms for pharmacogenomic variations and aggregating their predictions into a unified ensemble score. Importantly, APF2 provides quantitative variant effect estimates that correlate well with experimental results (R2 = 0.91, p = 0.003) and predicts the functional impact of pharmacogenomic variants with higher accuracy than previous methods, particularly for clinically relevant variations with actionable pharmacogenomic guidelines. We furthermore demonstrate better performance (92% accuracy) on an independent test set of 146 variants across 61 pharmacogenes not used for model training or validation. Application of APF2 to population-scale sequencing data from over 800,000 individuals revealed drastic ethnogeographic differences with important implications for pharmacotherapy. We thus think that APF2 holds the potential to improve the translation of genetic information into pharmacogenetic recommendations, thereby facilitating the use of Next-Generation Sequencing data for stratified medicine.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Pharmacogenomics Journal
医学-药学
CiteScore
7.20
自引率
0.00%
发文量
35
审稿时长
6-12 weeks
期刊介绍:
The Pharmacogenomics Journal is a print and electronic journal, which is dedicated to the rapid publication of original research on pharmacogenomics and its clinical applications.
Key areas of coverage include:
Personalized medicine
Effects of genetic variability on drug toxicity and efficacy
Identification and functional characterization of polymorphisms relevant to drug action
Pharmacodynamic and pharmacokinetic variations and drug efficacy
Integration of new developments in the genome project and proteomics into clinical medicine, pharmacology, and therapeutics
Clinical applications of genomic science
Identification of novel genomic targets for drug development
Potential benefits of pharmacogenomics.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


