Yunli Eric Hsieh, Kshitij Tandon, Heroen Verbruggen, Zoran Nikoloski
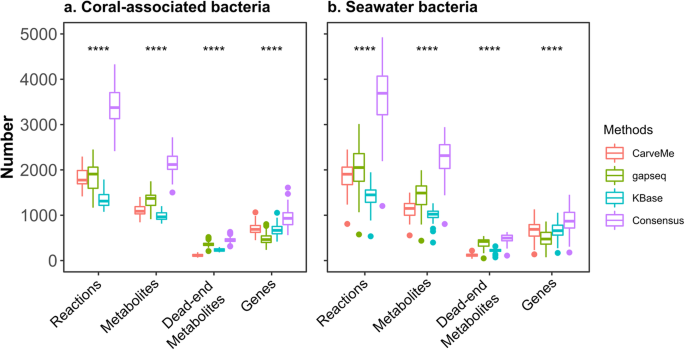
{"title":"通过自动工具和共识方法重建的微生物群落代谢模型的比较分析。","authors":"Yunli Eric Hsieh, Kshitij Tandon, Heroen Verbruggen, Zoran Nikoloski","doi":"10.1038/s41540-024-00384-y","DOIUrl":null,"url":null,"abstract":"<p><p>Genome-scale metabolic models (GEMs) of microbial communities offer valuable insights into the functional capabilities of their members and facilitate the exploration of microbial interactions. These models are generated using different automated reconstruction tools, each relying on different biochemical databases that may affect the conclusions drawn from the in silico analysis. One way to address this problem is to employ a consensus reconstruction method that combines the outcomes of different reconstruction tools. Here, we conducted a comparative analysis of community models reconstructed from three automated tools, i.e. CarveMe, gapseq, and KBase, alongside a consensus approach, utilizing metagenomics data from two marine bacterial communities. Our analysis revealed that these reconstruction approaches, while based on the same genomes, resulted in GEMs with varying numbers of genes and reactions as well as metabolic functionalities, attributed to the different databases employed. Further, our results indicated that the set of exchanged metabolites was more influenced by the reconstruction approach rather than the specific bacterial community investigated. This observation suggests a potential bias in predicting metabolite interactions using community GEMs. We also showed that consensus models encompassed a larger number of reactions and metabolites while concurrently reducing the presence of dead-end metabolites. Therefore, the usage of consensus models allows making full and unbiased use from aggregating genes from the different reconstructions in assessing the functional potential of microbial communities.</p>","PeriodicalId":19345,"journal":{"name":"NPJ Systems Biology and Applications","volume":null,"pages":null},"PeriodicalIF":3.5000,"publicationDate":"2024-05-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11116368/pdf/","citationCount":"0","resultStr":"{\"title\":\"Comparative analysis of metabolic models of microbial communities reconstructed from automated tools and consensus approaches.\",\"authors\":\"Yunli Eric Hsieh, Kshitij Tandon, Heroen Verbruggen, Zoran Nikoloski\",\"doi\":\"10.1038/s41540-024-00384-y\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Genome-scale metabolic models (GEMs) of microbial communities offer valuable insights into the functional capabilities of their members and facilitate the exploration of microbial interactions. These models are generated using different automated reconstruction tools, each relying on different biochemical databases that may affect the conclusions drawn from the in silico analysis. One way to address this problem is to employ a consensus reconstruction method that combines the outcomes of different reconstruction tools. Here, we conducted a comparative analysis of community models reconstructed from three automated tools, i.e. CarveMe, gapseq, and KBase, alongside a consensus approach, utilizing metagenomics data from two marine bacterial communities. Our analysis revealed that these reconstruction approaches, while based on the same genomes, resulted in GEMs with varying numbers of genes and reactions as well as metabolic functionalities, attributed to the different databases employed. Further, our results indicated that the set of exchanged metabolites was more influenced by the reconstruction approach rather than the specific bacterial community investigated. This observation suggests a potential bias in predicting metabolite interactions using community GEMs. We also showed that consensus models encompassed a larger number of reactions and metabolites while concurrently reducing the presence of dead-end metabolites. Therefore, the usage of consensus models allows making full and unbiased use from aggregating genes from the different reconstructions in assessing the functional potential of microbial communities.</p>\",\"PeriodicalId\":19345,\"journal\":{\"name\":\"NPJ Systems Biology and Applications\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":3.5000,\"publicationDate\":\"2024-05-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11116368/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"NPJ Systems Biology and Applications\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1038/s41540-024-00384-y\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"NPJ Systems Biology and Applications","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1038/s41540-024-00384-y","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
Comparative analysis of metabolic models of microbial communities reconstructed from automated tools and consensus approaches.
Genome-scale metabolic models (GEMs) of microbial communities offer valuable insights into the functional capabilities of their members and facilitate the exploration of microbial interactions. These models are generated using different automated reconstruction tools, each relying on different biochemical databases that may affect the conclusions drawn from the in silico analysis. One way to address this problem is to employ a consensus reconstruction method that combines the outcomes of different reconstruction tools. Here, we conducted a comparative analysis of community models reconstructed from three automated tools, i.e. CarveMe, gapseq, and KBase, alongside a consensus approach, utilizing metagenomics data from two marine bacterial communities. Our analysis revealed that these reconstruction approaches, while based on the same genomes, resulted in GEMs with varying numbers of genes and reactions as well as metabolic functionalities, attributed to the different databases employed. Further, our results indicated that the set of exchanged metabolites was more influenced by the reconstruction approach rather than the specific bacterial community investigated. This observation suggests a potential bias in predicting metabolite interactions using community GEMs. We also showed that consensus models encompassed a larger number of reactions and metabolites while concurrently reducing the presence of dead-end metabolites. Therefore, the usage of consensus models allows making full and unbiased use from aggregating genes from the different reconstructions in assessing the functional potential of microbial communities.
期刊介绍:
npj Systems Biology and Applications is an online Open Access journal dedicated to publishing the premier research that takes a systems-oriented approach. The journal aims to provide a forum for the presentation of articles that help define this nascent field, as well as those that apply the advances to wider fields. We encourage studies that integrate, or aid the integration of, data, analyses and insight from molecules to organisms and broader systems. Important areas of interest include not only fundamental biological systems and drug discovery, but also applications to health, medical practice and implementation, big data, biotechnology, food science, human behaviour, broader biological systems and industrial applications of systems biology.
We encourage all approaches, including network biology, application of control theory to biological systems, computational modelling and analysis, comprehensive and/or high-content measurements, theoretical, analytical and computational studies of system-level properties of biological systems and computational/software/data platforms enabling such studies.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


