Katarzyna Górczak , Tomasz Burzykowski , Jürgen Claesen
{"title":"用于分析甲基化测序数据的变化系数模型","authors":"Katarzyna Górczak , Tomasz Burzykowski , Jürgen Claesen","doi":"10.1016/j.compbiolchem.2024.108094","DOIUrl":null,"url":null,"abstract":"<div><p>DNA methylation is an important epigenetic modification involved in gene regulation. Advances in the next generation sequencing technology have enabled the retrieval of DNA methylation information at single-base-resolution. However, due to the sequencing process and the limited amount of isolated DNA, DNA-methylation-data are often noisy and sparse, which complicates the identification of differentially methylated regions (DMRs), especially when few replicates are available. We present a varying-coefficient model for detecting DMRs by using single-base-resolved methylation information. The model simultaneously smooths the methylation profiles and allows detection of DMRs, while accounting for additional covariates. The proposed model takes into account possible overdispersion by using a beta-binomial distribution. The overdispersion itself can be modeled as a function of the genomic region and explanatory variables. We illustrate the properties of the proposed model by applying it to two real-life case studies.</p></div>","PeriodicalId":10616,"journal":{"name":"Computational Biology and Chemistry","volume":null,"pages":null},"PeriodicalIF":2.6000,"publicationDate":"2024-05-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S1476927124000823/pdfft?md5=67c0caa95b43f4260e4b2a1c0f021788&pid=1-s2.0-S1476927124000823-main.pdf","citationCount":"0","resultStr":"{\"title\":\"A varying-coefficient model for the analysis of methylation sequencing data\",\"authors\":\"Katarzyna Górczak , Tomasz Burzykowski , Jürgen Claesen\",\"doi\":\"10.1016/j.compbiolchem.2024.108094\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>DNA methylation is an important epigenetic modification involved in gene regulation. Advances in the next generation sequencing technology have enabled the retrieval of DNA methylation information at single-base-resolution. However, due to the sequencing process and the limited amount of isolated DNA, DNA-methylation-data are often noisy and sparse, which complicates the identification of differentially methylated regions (DMRs), especially when few replicates are available. We present a varying-coefficient model for detecting DMRs by using single-base-resolved methylation information. The model simultaneously smooths the methylation profiles and allows detection of DMRs, while accounting for additional covariates. The proposed model takes into account possible overdispersion by using a beta-binomial distribution. The overdispersion itself can be modeled as a function of the genomic region and explanatory variables. We illustrate the properties of the proposed model by applying it to two real-life case studies.</p></div>\",\"PeriodicalId\":10616,\"journal\":{\"name\":\"Computational Biology and Chemistry\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":2.6000,\"publicationDate\":\"2024-05-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.sciencedirect.com/science/article/pii/S1476927124000823/pdfft?md5=67c0caa95b43f4260e4b2a1c0f021788&pid=1-s2.0-S1476927124000823-main.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Computational Biology and Chemistry\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S1476927124000823\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational Biology and Chemistry","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1476927124000823","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
DNA 甲基化是参与基因调控的重要表观遗传修饰。新一代测序技术的进步使人们能够以单碱基分辨率检索 DNA 甲基化信息。然而,由于测序过程和分离出的 DNA 数量有限,DNA 甲基化数据往往是嘈杂和稀疏的,这使得差异甲基化区域(DMR)的鉴定变得复杂,尤其是在只有少量重复数据的情况下。我们利用单碱基分辨甲基化信息提出了一种检测 DMR 的变化系数模型。该模型可同时平滑甲基化图谱并检测 DMR,同时考虑额外的协变量。所提出的模型通过使用贝塔二叉分布,考虑到了可能出现的过度分散。过度分散本身可以作为基因组区域和解释变量的函数来建模。我们将拟议模型应用于两个实际案例研究,以说明该模型的特性。
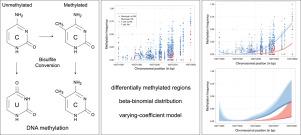
A varying-coefficient model for the analysis of methylation sequencing data
DNA methylation is an important epigenetic modification involved in gene regulation. Advances in the next generation sequencing technology have enabled the retrieval of DNA methylation information at single-base-resolution. However, due to the sequencing process and the limited amount of isolated DNA, DNA-methylation-data are often noisy and sparse, which complicates the identification of differentially methylated regions (DMRs), especially when few replicates are available. We present a varying-coefficient model for detecting DMRs by using single-base-resolved methylation information. The model simultaneously smooths the methylation profiles and allows detection of DMRs, while accounting for additional covariates. The proposed model takes into account possible overdispersion by using a beta-binomial distribution. The overdispersion itself can be modeled as a function of the genomic region and explanatory variables. We illustrate the properties of the proposed model by applying it to two real-life case studies.
期刊介绍:
Computational Biology and Chemistry publishes original research papers and review articles in all areas of computational life sciences. High quality research contributions with a major computational component in the areas of nucleic acid and protein sequence research, molecular evolution, molecular genetics (functional genomics and proteomics), theory and practice of either biology-specific or chemical-biology-specific modeling, and structural biology of nucleic acids and proteins are particularly welcome. Exceptionally high quality research work in bioinformatics, systems biology, ecology, computational pharmacology, metabolism, biomedical engineering, epidemiology, and statistical genetics will also be considered.
Given their inherent uncertainty, protein modeling and molecular docking studies should be thoroughly validated. In the absence of experimental results for validation, the use of molecular dynamics simulations along with detailed free energy calculations, for example, should be used as complementary techniques to support the major conclusions. Submissions of premature modeling exercises without additional biological insights will not be considered.
Review articles will generally be commissioned by the editors and should not be submitted to the journal without explicit invitation. However prospective authors are welcome to send a brief (one to three pages) synopsis, which will be evaluated by the editors.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


