Jiayi Xu , Jianting Gong , Xiaochen Bo , Yigang Tong , Zilin Ren , Ming Ni
{"title":"基于结构的抗体-抗原结合亲和力在线工具评估基准","authors":"Jiayi Xu , Jianting Gong , Xiaochen Bo , Yigang Tong , Zilin Ren , Ming Ni","doi":"10.1016/j.bpc.2024.107253","DOIUrl":null,"url":null,"abstract":"<div><p>The prediction of binding affinity changes caused by missense mutations can elucidate antigen-antibody interactions. A few accessible structure-based online computational tools have been proposed. However, selecting suitable software for particular research is challenging, especially research on the SARS-CoV-2 spike protein with antibodies. Therefore, benchmarking of the mutation-diverse SARS-CoV-2 datasets is critical. Here, we collected the datasets including 1216 variants about the changes in binding affinity of antigens from 22 complexes for SARS-CoV-2 S proteins and 22 monoclonal antibodies as well as applied them to evaluate the performance of seven binding affinity prediction tools. The tested tools' Pearson correlations between predicted and measured changes in binding affinity were between −0.158 and 0.657, while accuracy in classification tasks on predicting increasing or decreasing affinity ranged from 0.444 to 0.834. These tools performed relatively better on predicting single mutations, especially at epitope sites, whereas poor performance on extremely decreasing affinity. The tested tools were relatively insensitive to the experimental techniques used to obtain structures of complexes. In summary, we constructed a list of datasets and evaluated a range of structure-based online prediction tools that will explicate relevant processes of antigen-antibody interactions and enhance the computational design of therapeutic monoclonal antibodies.</p></div>","PeriodicalId":8979,"journal":{"name":"Biophysical chemistry","volume":"311 ","pages":"Article 107253"},"PeriodicalIF":3.3000,"publicationDate":"2024-04-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"A benchmark for evaluation of structure-based online tools for antibody-antigen binding affinity\",\"authors\":\"Jiayi Xu , Jianting Gong , Xiaochen Bo , Yigang Tong , Zilin Ren , Ming Ni\",\"doi\":\"10.1016/j.bpc.2024.107253\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>The prediction of binding affinity changes caused by missense mutations can elucidate antigen-antibody interactions. A few accessible structure-based online computational tools have been proposed. However, selecting suitable software for particular research is challenging, especially research on the SARS-CoV-2 spike protein with antibodies. Therefore, benchmarking of the mutation-diverse SARS-CoV-2 datasets is critical. Here, we collected the datasets including 1216 variants about the changes in binding affinity of antigens from 22 complexes for SARS-CoV-2 S proteins and 22 monoclonal antibodies as well as applied them to evaluate the performance of seven binding affinity prediction tools. The tested tools' Pearson correlations between predicted and measured changes in binding affinity were between −0.158 and 0.657, while accuracy in classification tasks on predicting increasing or decreasing affinity ranged from 0.444 to 0.834. These tools performed relatively better on predicting single mutations, especially at epitope sites, whereas poor performance on extremely decreasing affinity. The tested tools were relatively insensitive to the experimental techniques used to obtain structures of complexes. In summary, we constructed a list of datasets and evaluated a range of structure-based online prediction tools that will explicate relevant processes of antigen-antibody interactions and enhance the computational design of therapeutic monoclonal antibodies.</p></div>\",\"PeriodicalId\":8979,\"journal\":{\"name\":\"Biophysical chemistry\",\"volume\":\"311 \",\"pages\":\"Article 107253\"},\"PeriodicalIF\":3.3000,\"publicationDate\":\"2024-04-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Biophysical chemistry\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0301462224000826\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biophysical chemistry","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0301462224000826","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
A benchmark for evaluation of structure-based online tools for antibody-antigen binding affinity
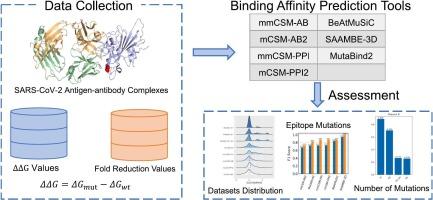
The prediction of binding affinity changes caused by missense mutations can elucidate antigen-antibody interactions. A few accessible structure-based online computational tools have been proposed. However, selecting suitable software for particular research is challenging, especially research on the SARS-CoV-2 spike protein with antibodies. Therefore, benchmarking of the mutation-diverse SARS-CoV-2 datasets is critical. Here, we collected the datasets including 1216 variants about the changes in binding affinity of antigens from 22 complexes for SARS-CoV-2 S proteins and 22 monoclonal antibodies as well as applied them to evaluate the performance of seven binding affinity prediction tools. The tested tools' Pearson correlations between predicted and measured changes in binding affinity were between −0.158 and 0.657, while accuracy in classification tasks on predicting increasing or decreasing affinity ranged from 0.444 to 0.834. These tools performed relatively better on predicting single mutations, especially at epitope sites, whereas poor performance on extremely decreasing affinity. The tested tools were relatively insensitive to the experimental techniques used to obtain structures of complexes. In summary, we constructed a list of datasets and evaluated a range of structure-based online prediction tools that will explicate relevant processes of antigen-antibody interactions and enhance the computational design of therapeutic monoclonal antibodies.
期刊介绍:
Biophysical Chemistry publishes original work and reviews in the areas of chemistry and physics directly impacting biological phenomena. Quantitative analysis of the properties of biological macromolecules, biologically active molecules, macromolecular assemblies and cell components in terms of kinetics, thermodynamics, spatio-temporal organization, NMR and X-ray structural biology, as well as single-molecule detection represent a major focus of the journal. Theoretical and computational treatments of biomacromolecular systems, macromolecular interactions, regulatory control and systems biology are also of interest to the journal.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


