{"title":"利用联合稀疏典型相关分析确定与疾病结果相关的基因--在肾透明细胞癌中的应用","authors":"Diptavo Dutta, Ananda Sen, Jaya M. Satagopan","doi":"10.1002/gepi.22566","DOIUrl":null,"url":null,"abstract":"<p>Somatic changes like copy number aberrations (CNAs) and epigenetic alterations like methylation have pivotal effects on disease outcomes and prognosis in cancer, by regulating gene expressions, that drive critical biological processes. To identify potential biomarkers and molecular targets and understand how they impact disease outcomes, it is important to identify key groups of CNAs, the associated methylation, and the gene expressions they impact, through a joint integrative analysis. Here, we propose a novel analysis pipeline, the joint sparse canonical correlation analysis (jsCCA), an extension of sCCA, to effectively identify an ensemble of CNAs, methylation sites and gene (expression) components in the context of disease endpoints, especially tumor characteristics. Our approach detects potentially orthogonal gene components that are highly correlated with sets of methylation sites which in turn are correlated with sets of CNA sites. It then identifies the genes within these components that are associated with the outcome. Further, we aggregate the effect of each gene expression set on tumor stage by constructing “gene component scores” and test its interaction with traditional risk factors. Analyzing clinical and genomic data on 515 renal clear cell carcinoma (ccRCC) patients from the TCGA-KIRC, we found eight gene components to be associated with methylation sites, regulated by groups of proximally located CNA sites. Association analysis with tumor stage at diagnosis identified a novel association of expression of <i>ASAH1</i> gene trans-regulated by methylation of several genes including <i>SIX5</i> and by CNAs in the 10q25 region including <i>TCF7L2</i>. Further analysis to quantify the overall effect of gene sets on tumor stage, revealed that two of the eight gene components have significant interaction with smoking in relation to tumor stage. These gene components represent distinct biological functions including immune function, inflammatory responses, and hypoxia-regulated pathways. Our findings suggest that jsCCA analysis can identify interpretable and important genes, regulatory structures, and clinically consequential pathways. Such methods are warranted for comprehensive analysis of multimodal data especially in cancer genomics.</p>","PeriodicalId":12710,"journal":{"name":"Genetic Epidemiology","volume":"48 8","pages":"414-432"},"PeriodicalIF":3.8000,"publicationDate":"2024-05-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/gepi.22566","citationCount":"0","resultStr":"{\"title\":\"Identifying genes associated with disease outcomes using joint sparse canonical correlation analysis—An application in renal clear cell carcinoma\",\"authors\":\"Diptavo Dutta, Ananda Sen, Jaya M. Satagopan\",\"doi\":\"10.1002/gepi.22566\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Somatic changes like copy number aberrations (CNAs) and epigenetic alterations like methylation have pivotal effects on disease outcomes and prognosis in cancer, by regulating gene expressions, that drive critical biological processes. To identify potential biomarkers and molecular targets and understand how they impact disease outcomes, it is important to identify key groups of CNAs, the associated methylation, and the gene expressions they impact, through a joint integrative analysis. Here, we propose a novel analysis pipeline, the joint sparse canonical correlation analysis (jsCCA), an extension of sCCA, to effectively identify an ensemble of CNAs, methylation sites and gene (expression) components in the context of disease endpoints, especially tumor characteristics. Our approach detects potentially orthogonal gene components that are highly correlated with sets of methylation sites which in turn are correlated with sets of CNA sites. It then identifies the genes within these components that are associated with the outcome. Further, we aggregate the effect of each gene expression set on tumor stage by constructing “gene component scores” and test its interaction with traditional risk factors. Analyzing clinical and genomic data on 515 renal clear cell carcinoma (ccRCC) patients from the TCGA-KIRC, we found eight gene components to be associated with methylation sites, regulated by groups of proximally located CNA sites. Association analysis with tumor stage at diagnosis identified a novel association of expression of <i>ASAH1</i> gene trans-regulated by methylation of several genes including <i>SIX5</i> and by CNAs in the 10q25 region including <i>TCF7L2</i>. Further analysis to quantify the overall effect of gene sets on tumor stage, revealed that two of the eight gene components have significant interaction with smoking in relation to tumor stage. These gene components represent distinct biological functions including immune function, inflammatory responses, and hypoxia-regulated pathways. Our findings suggest that jsCCA analysis can identify interpretable and important genes, regulatory structures, and clinically consequential pathways. Such methods are warranted for comprehensive analysis of multimodal data especially in cancer genomics.</p>\",\"PeriodicalId\":12710,\"journal\":{\"name\":\"Genetic Epidemiology\",\"volume\":\"48 8\",\"pages\":\"414-432\"},\"PeriodicalIF\":3.8000,\"publicationDate\":\"2024-05-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/gepi.22566\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Genetic Epidemiology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/gepi.22566\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genetic Epidemiology","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/gepi.22566","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Identifying genes associated with disease outcomes using joint sparse canonical correlation analysis—An application in renal clear cell carcinoma
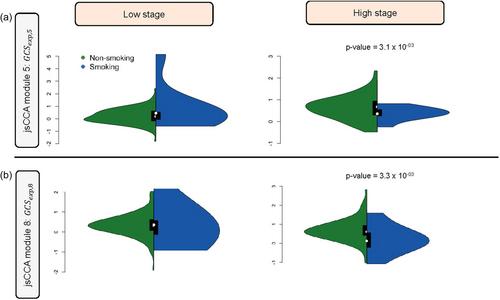
Somatic changes like copy number aberrations (CNAs) and epigenetic alterations like methylation have pivotal effects on disease outcomes and prognosis in cancer, by regulating gene expressions, that drive critical biological processes. To identify potential biomarkers and molecular targets and understand how they impact disease outcomes, it is important to identify key groups of CNAs, the associated methylation, and the gene expressions they impact, through a joint integrative analysis. Here, we propose a novel analysis pipeline, the joint sparse canonical correlation analysis (jsCCA), an extension of sCCA, to effectively identify an ensemble of CNAs, methylation sites and gene (expression) components in the context of disease endpoints, especially tumor characteristics. Our approach detects potentially orthogonal gene components that are highly correlated with sets of methylation sites which in turn are correlated with sets of CNA sites. It then identifies the genes within these components that are associated with the outcome. Further, we aggregate the effect of each gene expression set on tumor stage by constructing “gene component scores” and test its interaction with traditional risk factors. Analyzing clinical and genomic data on 515 renal clear cell carcinoma (ccRCC) patients from the TCGA-KIRC, we found eight gene components to be associated with methylation sites, regulated by groups of proximally located CNA sites. Association analysis with tumor stage at diagnosis identified a novel association of expression of ASAH1 gene trans-regulated by methylation of several genes including SIX5 and by CNAs in the 10q25 region including TCF7L2. Further analysis to quantify the overall effect of gene sets on tumor stage, revealed that two of the eight gene components have significant interaction with smoking in relation to tumor stage. These gene components represent distinct biological functions including immune function, inflammatory responses, and hypoxia-regulated pathways. Our findings suggest that jsCCA analysis can identify interpretable and important genes, regulatory structures, and clinically consequential pathways. Such methods are warranted for comprehensive analysis of multimodal data especially in cancer genomics.
期刊介绍:
Genetic Epidemiology is a peer-reviewed journal for discussion of research on the genetic causes of the distribution of human traits in families and populations. Emphasis is placed on the relative contribution of genetic and environmental factors to human disease as revealed by genetic, epidemiological, and biologic investigations.
Genetic Epidemiology primarily publishes papers in statistical genetics, a research field that is primarily concerned with development of statistical, bioinformatical, and computational models for analyzing genetic data. Incorporation of underlying biology and population genetics into conceptual models is favored. The Journal seeks original articles comprising either applied research or innovative statistical, mathematical, computational, or genomic methodologies that advance studies in genetic epidemiology. Other types of reports are encouraged, such as letters to the editor, topic reviews, and perspectives from other fields of research that will likely enrich the field of genetic epidemiology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


