{"title":"了解细菌病原体的多样性:蛋白质基因组分析和基因组组装阵列的使用,以确定蜜蜂细菌病原体幼虫担子菌的新型毒力因子。","authors":"Tomas Erban, Bruno Sopko","doi":"10.1002/pmic.202300280","DOIUrl":null,"url":null,"abstract":"<p>Mass spectrometry proteomics data are typically evaluated against publicly available annotated sequences, but the proteogenomics approach is a useful alternative. A single genome is commonly utilized in custom proteomic and proteogenomic data analysis. We pose the question of whether utilizing numerous different genome assemblies in a search database would be beneficial. We reanalyzed raw data from the exoprotein fraction of four reference Enterobacterial Repetitive Intergenic Consensus (ERIC) I–IV genotypes of the honey bee bacterial pathogen <i>Paenibacillus larvae</i> and evaluated them against three reference databases (from NCBI-protein, RefSeq, and UniProt) together with an array of protein sequences generated by six-frame direct translation of 15 genome assemblies from GenBank. The wide search yielded 453 protein hits/groups, which UpSet analysis categorized into 50 groups based on the success of protein identification by the 18 database components. Nine hits that were not identified by a unique peptide were not considered for marker selection, which discarded the only protein that was not identified by the reference databases. We propose that the variability in successful identifications between genome assemblies is useful for marker mining. The results suggest that various strains of <i>P. larvae</i> can exhibit specific traits that set them apart from the established genotypes ERIC I–V.</p>","PeriodicalId":224,"journal":{"name":"Proteomics","volume":"24 14","pages":""},"PeriodicalIF":3.4000,"publicationDate":"2024-05-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/pmic.202300280","citationCount":"0","resultStr":"{\"title\":\"Understanding bacterial pathogen diversity: A proteogenomic analysis and use of an array of genome assemblies to identify novel virulence factors of the honey bee bacterial pathogen Paenibacillus larvae\",\"authors\":\"Tomas Erban, Bruno Sopko\",\"doi\":\"10.1002/pmic.202300280\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Mass spectrometry proteomics data are typically evaluated against publicly available annotated sequences, but the proteogenomics approach is a useful alternative. A single genome is commonly utilized in custom proteomic and proteogenomic data analysis. We pose the question of whether utilizing numerous different genome assemblies in a search database would be beneficial. We reanalyzed raw data from the exoprotein fraction of four reference Enterobacterial Repetitive Intergenic Consensus (ERIC) I–IV genotypes of the honey bee bacterial pathogen <i>Paenibacillus larvae</i> and evaluated them against three reference databases (from NCBI-protein, RefSeq, and UniProt) together with an array of protein sequences generated by six-frame direct translation of 15 genome assemblies from GenBank. The wide search yielded 453 protein hits/groups, which UpSet analysis categorized into 50 groups based on the success of protein identification by the 18 database components. Nine hits that were not identified by a unique peptide were not considered for marker selection, which discarded the only protein that was not identified by the reference databases. We propose that the variability in successful identifications between genome assemblies is useful for marker mining. The results suggest that various strains of <i>P. larvae</i> can exhibit specific traits that set them apart from the established genotypes ERIC I–V.</p>\",\"PeriodicalId\":224,\"journal\":{\"name\":\"Proteomics\",\"volume\":\"24 14\",\"pages\":\"\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2024-05-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/pmic.202300280\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Proteomics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/pmic.202300280\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Proteomics","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/pmic.202300280","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
摘要
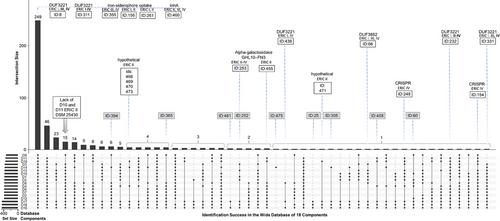
质谱蛋白质组学数据通常根据公开的注释序列进行评估,但蛋白质基因组学方法是一种有用的替代方法。在定制蛋白质组学和蛋白质基因组学数据分析中,通常使用单一基因组。我们提出的问题是,在搜索数据库中利用多个不同的基因组组装是否有益。我们重新分析了蜜蜂细菌病原体幼虫Paenibacillus的四种参考肠杆菌重复基因间共识(ERIC)I-IV基因型的外显子蛋白部分的原始数据,并对照三个参考数据库(NCBI-protein、RefSeq和UniProt)以及由GenBank中15个基因组组装的六帧直接翻译生成的蛋白质序列阵列进行了评估。根据 18 个数据库组件对蛋白质识别的成功率,UpSet 分析将其分为 50 组。在标记选择时,9 个未被唯一肽鉴定的点击未被考虑,这就摒弃了唯一一个未被参考数据库鉴定的蛋白质。我们认为,不同基因组组装之间成功鉴定的差异有助于标记挖掘。结果表明,各种幼虫品系都能表现出特定的性状,使其有别于已建立的基因型 ERIC I-V。
Understanding bacterial pathogen diversity: A proteogenomic analysis and use of an array of genome assemblies to identify novel virulence factors of the honey bee bacterial pathogen Paenibacillus larvae
Mass spectrometry proteomics data are typically evaluated against publicly available annotated sequences, but the proteogenomics approach is a useful alternative. A single genome is commonly utilized in custom proteomic and proteogenomic data analysis. We pose the question of whether utilizing numerous different genome assemblies in a search database would be beneficial. We reanalyzed raw data from the exoprotein fraction of four reference Enterobacterial Repetitive Intergenic Consensus (ERIC) I–IV genotypes of the honey bee bacterial pathogen Paenibacillus larvae and evaluated them against three reference databases (from NCBI-protein, RefSeq, and UniProt) together with an array of protein sequences generated by six-frame direct translation of 15 genome assemblies from GenBank. The wide search yielded 453 protein hits/groups, which UpSet analysis categorized into 50 groups based on the success of protein identification by the 18 database components. Nine hits that were not identified by a unique peptide were not considered for marker selection, which discarded the only protein that was not identified by the reference databases. We propose that the variability in successful identifications between genome assemblies is useful for marker mining. The results suggest that various strains of P. larvae can exhibit specific traits that set them apart from the established genotypes ERIC I–V.
期刊介绍:
PROTEOMICS is the premier international source for information on all aspects of applications and technologies, including software, in proteomics and other "omics". The journal includes but is not limited to proteomics, genomics, transcriptomics, metabolomics and lipidomics, and systems biology approaches. Papers describing novel applications of proteomics and integration of multi-omics data and approaches are especially welcome.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


