Fulin Mao , Weizhong Shi , Hongkun Zhao , Wanxin Li , Abolghasem Jouyban , Ali Farajtabar
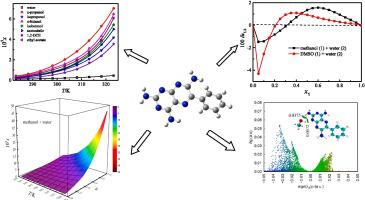
{"title":"根据溶剂效应、模型、热力学分析和量子化学计算研究三苯酯在单溶剂和水溶液中的平衡溶解度","authors":"Fulin Mao , Weizhong Shi , Hongkun Zhao , Wanxin Li , Abolghasem Jouyban , Ali Farajtabar","doi":"10.1016/j.jct.2024.107324","DOIUrl":null,"url":null,"abstract":"<div><p>The equilibrium solubilities of triamterene in pure solvents {isopropanol, methanol, <em>n</em>-propanol, water, 1,2-dichloroethane, 2-methoxyethanol, ethyl acetate, acetonitrile, <em>N</em>-methylformamide (NMF), <em>n</em>-butanol, isobutanol and dimethylsulfoxide (DMSO)}, as well as solvent mixtures of methanol/DMSO + water, were determined by dint of the saturation shake-flask technique. Triamterene solubility (10<sup>5</sup><em>x</em>) rose as the determination temperature rose and fell in the order at 298.15 K: 2-methoxyethanol (453.9) > DMSO (281.3) > NMF (142.6) > methanol (8.424) > <em>n</em>-propanol (1.432) > <em>n</em>-butanol (1.152) > ethyl acetate (0.9441) > isobutanol (0.8738) > acetonitrile (0.7991) > isopropanol (0.7419) > 1,2-dichloroethane (0.5232) > water (0.1795). No crystal transition or solvation in the trial process was shown via X -ray power diffraction patterns. The solvent effects, in which the interactions of solvent–solvent and solvent–solute were considered, were studied via linear solvation energy relationship. Using equations such as Wilson, Apelblat, <em>λh</em>, and NRTL for mono-solvents and Jouyban-Acree as well as modified van't Hoff-Jouyban-Acree for solvent mixtures, the magnitudes of equilibrium solubility were associated. The greatest relative average deviation of 5.787 × 10<sup>−2</sup> and the maximum deviation value of root mean square of 219.1 × 10<sup>−4</sup> were computed for neat solvents. According to the solubility information in pure solvents, the Wilson equation was used to compute the mixing solution parameters. At 298.15 K, the inverse Kirkwood-Buff integral was utilized here to examine the preferred solvation of triamterene. In blends of DMSO/methanol + water with middle and rich DMSO/methanol compositions, the parameters of preferred solvation for DMSO/methanol were positive, indicating the preferential solvation of triamterene by DMSO/methanol. In addition, the electrostatic acidity-basicity properties were demonstrated using the lowest negative electrostatic potential in company with the minimum m local ionization energy of molecular surface. The <img>N<img> in ring of the triamterene is the preferential target for electrophilic assault; and –NH<sub>2</sub> of the triamterene, for nucleophilic assault. The weak triamterene-solvent interactions were graphically displayed by the use of the independent gradient approach on the basis of Hirshfeld partition analysis.</p></div>","PeriodicalId":54867,"journal":{"name":"Journal of Chemical Thermodynamics","volume":"196 ","pages":"Article 107324"},"PeriodicalIF":2.2000,"publicationDate":"2024-05-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Research on triamterene equilibrium solubility in mono-solvents and aqueous solutions according to solvent effect, models, thermodynamic analysis and quantum chemical calculation\",\"authors\":\"Fulin Mao , Weizhong Shi , Hongkun Zhao , Wanxin Li , Abolghasem Jouyban , Ali Farajtabar\",\"doi\":\"10.1016/j.jct.2024.107324\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>The equilibrium solubilities of triamterene in pure solvents {isopropanol, methanol, <em>n</em>-propanol, water, 1,2-dichloroethane, 2-methoxyethanol, ethyl acetate, acetonitrile, <em>N</em>-methylformamide (NMF), <em>n</em>-butanol, isobutanol and dimethylsulfoxide (DMSO)}, as well as solvent mixtures of methanol/DMSO + water, were determined by dint of the saturation shake-flask technique. Triamterene solubility (10<sup>5</sup><em>x</em>) rose as the determination temperature rose and fell in the order at 298.15 K: 2-methoxyethanol (453.9) > DMSO (281.3) > NMF (142.6) > methanol (8.424) > <em>n</em>-propanol (1.432) > <em>n</em>-butanol (1.152) > ethyl acetate (0.9441) > isobutanol (0.8738) > acetonitrile (0.7991) > isopropanol (0.7419) > 1,2-dichloroethane (0.5232) > water (0.1795). No crystal transition or solvation in the trial process was shown via X -ray power diffraction patterns. The solvent effects, in which the interactions of solvent–solvent and solvent–solute were considered, were studied via linear solvation energy relationship. Using equations such as Wilson, Apelblat, <em>λh</em>, and NRTL for mono-solvents and Jouyban-Acree as well as modified van't Hoff-Jouyban-Acree for solvent mixtures, the magnitudes of equilibrium solubility were associated. The greatest relative average deviation of 5.787 × 10<sup>−2</sup> and the maximum deviation value of root mean square of 219.1 × 10<sup>−4</sup> were computed for neat solvents. According to the solubility information in pure solvents, the Wilson equation was used to compute the mixing solution parameters. At 298.15 K, the inverse Kirkwood-Buff integral was utilized here to examine the preferred solvation of triamterene. In blends of DMSO/methanol + water with middle and rich DMSO/methanol compositions, the parameters of preferred solvation for DMSO/methanol were positive, indicating the preferential solvation of triamterene by DMSO/methanol. In addition, the electrostatic acidity-basicity properties were demonstrated using the lowest negative electrostatic potential in company with the minimum m local ionization energy of molecular surface. The <img>N<img> in ring of the triamterene is the preferential target for electrophilic assault; and –NH<sub>2</sub> of the triamterene, for nucleophilic assault. The weak triamterene-solvent interactions were graphically displayed by the use of the independent gradient approach on the basis of Hirshfeld partition analysis.</p></div>\",\"PeriodicalId\":54867,\"journal\":{\"name\":\"Journal of Chemical Thermodynamics\",\"volume\":\"196 \",\"pages\":\"Article 107324\"},\"PeriodicalIF\":2.2000,\"publicationDate\":\"2024-05-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Thermodynamics\",\"FirstCategoryId\":\"5\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0021961424000776\",\"RegionNum\":3,\"RegionCategory\":\"工程技术\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Thermodynamics","FirstCategoryId":"5","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0021961424000776","RegionNum":3,"RegionCategory":"工程技术","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
采用饱和振荡烧瓶技术测定了三苯氧胺在纯溶剂{异丙醇、甲醇、正丙醇、水、1,2-二氯乙烷、2-甲氧基乙醇、乙酸乙酯、乙腈、N-甲基甲酰胺(NMF)、正丁醇、异丁醇和二甲基亚砜(DMSO)}以及甲醇/DMSO+水的混合溶剂中的平衡溶解度。三苯酯的溶解度(105x)随着测定温度的升高而升高,在 298.15 K 时依次为:2-甲氧基乙醇(453.9);二甲基亚砜(281.3);NMF(142.6);甲醇(8.424);正丙醇(1.432);正丁醇(1.152);乙酸乙酯(0.9441);异丁醇(0.8738);乙腈(0.7991);异丙醇(0.7419);1,2-二氯乙烷(0.5232);水(0.1795)。X 射线功率衍射图样显示,试验过程中没有晶体转变或溶解现象。通过线性溶解能关系研究了溶剂效应,其中考虑了溶剂-溶剂和溶剂-溶质之间的相互作用。对于单溶剂,使用 Wilson、Apelblat、λh 和 NRTL 等方程式;对于混合溶剂,使用 Jouyban-Acree 以及修正的 van't Hoff-Jouyban-Acree 等方程式,将平衡溶解度的大小联系起来。计算得出纯溶剂的最大相对平均偏差为 5.787 × 10-2,均方根最大偏差值为 219.1 × 10-4。根据纯溶剂中的溶解度信息,采用威尔逊方程计算混合溶液参数。在 298.15 K 的温度下,利用反 Kirkwood-Buff 积分来研究三苯氧胺的优先溶解度。在二甲基亚砜/甲醇+水的混合溶液中,中等和富含二甲基亚砜/甲醇的混合溶液中,二甲基亚砜/甲醇的优先溶解参数为正值,表明三苯氧胺优先溶解于二甲基亚砜/甲醇。此外,静电酸碱性特性是利用最低负静电位与分子表面最小 m 局域电离能共同作用来证明的。三苯氧胺环中的 N 是亲电攻击的首选目标;三苯氧胺的 -NH2 则是亲核攻击的首选目标。在 Hirshfeld 分配分析的基础上,利用独立梯度法以图形显示了三苯酯与溶剂之间的微弱相互作用。
Research on triamterene equilibrium solubility in mono-solvents and aqueous solutions according to solvent effect, models, thermodynamic analysis and quantum chemical calculation
The equilibrium solubilities of triamterene in pure solvents {isopropanol, methanol, n-propanol, water, 1,2-dichloroethane, 2-methoxyethanol, ethyl acetate, acetonitrile, N-methylformamide (NMF), n-butanol, isobutanol and dimethylsulfoxide (DMSO)}, as well as solvent mixtures of methanol/DMSO + water, were determined by dint of the saturation shake-flask technique. Triamterene solubility (105x) rose as the determination temperature rose and fell in the order at 298.15 K: 2-methoxyethanol (453.9) > DMSO (281.3) > NMF (142.6) > methanol (8.424) > n-propanol (1.432) > n-butanol (1.152) > ethyl acetate (0.9441) > isobutanol (0.8738) > acetonitrile (0.7991) > isopropanol (0.7419) > 1,2-dichloroethane (0.5232) > water (0.1795). No crystal transition or solvation in the trial process was shown via X -ray power diffraction patterns. The solvent effects, in which the interactions of solvent–solvent and solvent–solute were considered, were studied via linear solvation energy relationship. Using equations such as Wilson, Apelblat, λh, and NRTL for mono-solvents and Jouyban-Acree as well as modified van't Hoff-Jouyban-Acree for solvent mixtures, the magnitudes of equilibrium solubility were associated. The greatest relative average deviation of 5.787 × 10−2 and the maximum deviation value of root mean square of 219.1 × 10−4 were computed for neat solvents. According to the solubility information in pure solvents, the Wilson equation was used to compute the mixing solution parameters. At 298.15 K, the inverse Kirkwood-Buff integral was utilized here to examine the preferred solvation of triamterene. In blends of DMSO/methanol + water with middle and rich DMSO/methanol compositions, the parameters of preferred solvation for DMSO/methanol were positive, indicating the preferential solvation of triamterene by DMSO/methanol. In addition, the electrostatic acidity-basicity properties were demonstrated using the lowest negative electrostatic potential in company with the minimum m local ionization energy of molecular surface. The N in ring of the triamterene is the preferential target for electrophilic assault; and –NH2 of the triamterene, for nucleophilic assault. The weak triamterene-solvent interactions were graphically displayed by the use of the independent gradient approach on the basis of Hirshfeld partition analysis.
期刊介绍:
The Journal of Chemical Thermodynamics exists primarily for dissemination of significant new knowledge in experimental equilibrium thermodynamics and transport properties of chemical systems. The defining attributes of The Journal are the quality and relevance of the papers published.
The Journal publishes work relating to gases, liquids, solids, polymers, mixtures, solutions and interfaces. Studies on systems with variability, such as biological or bio-based materials, gas hydrates, among others, will also be considered provided these are well characterized and reproducible where possible. Experimental methods should be described in sufficient detail to allow critical assessment of the accuracy claimed.
Authors are encouraged to provide physical or chemical interpretations of the results. Articles can contain modelling sections providing representations of data or molecular insights into the properties or transformations studied. Theoretical papers on chemical thermodynamics using molecular theory or modelling are also considered.
The Journal welcomes review articles in the field of chemical thermodynamics but prospective authors should first consult one of the Editors concerning the suitability of the proposed review.
Contributions of a routine nature or reporting on uncharacterised materials are not accepted.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


