用深度学习预测分子系统的平衡分布
IF 18.8
1区 计算机科学
Q1 COMPUTER SCIENCE, ARTIFICIAL INTELLIGENCE
引用次数: 0
摘要
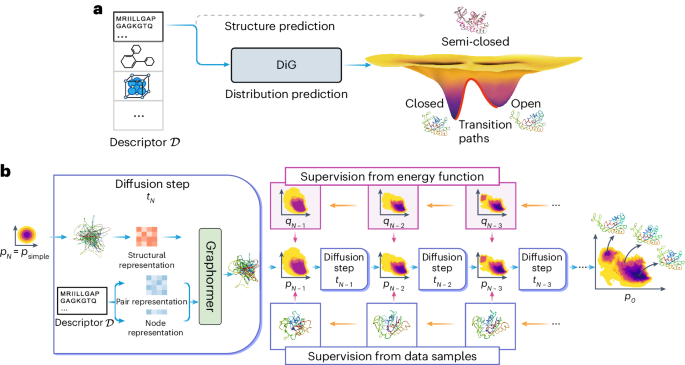
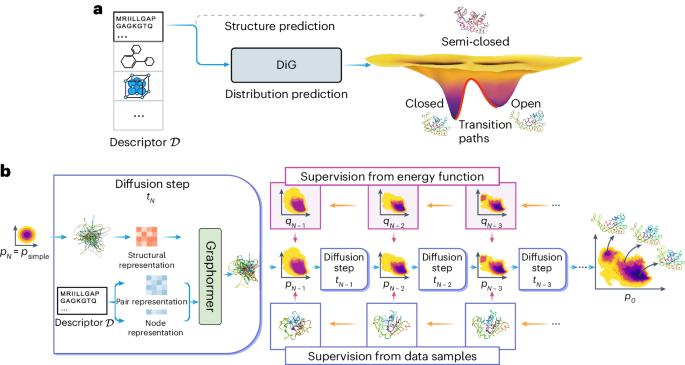
深度学习的进步极大地改进了分子结构预测。然而,对现实世界应用非常重要的许多宏观观测结果并不是单一分子结构的函数,而是由结构的平衡分布决定的。获取这些分布的传统方法(如分子动力学模拟)计算成本高昂,而且往往难以实现。在这里,我们引入了一个深度学习框架,称为分布式图解器(DiG),试图预测分子系统的平衡分布。受热力学退火过程的启发,DiG 利用深度神经网络将简单分布转化为平衡分布,并以分子系统的描述符(如化学图谱或蛋白质序列)为条件。这一框架能高效生成各种构象,并提供状态密度的估计值,其速度比传统方法快了几个数量级。我们展示了 DiG 在多个分子任务中的应用,包括蛋白质构象采样、配体结构采样、催化剂吸附剂采样和属性引导结构生成。DiG 在统计理解分子系统的方法上取得了重大进步,为分子科学开辟了新的研究机会。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Predicting equilibrium distributions for molecular systems with deep learning
Advances in deep learning have greatly improved structure prediction of molecules. However, many macroscopic observations that are important for real-world applications are not functions of a single molecular structure but rather determined from the equilibrium distribution of structures. Conventional methods for obtaining these distributions, such as molecular dynamics simulation, are computationally expensive and often intractable. Here we introduce a deep learning framework, called Distributional Graphormer (DiG), in an attempt to predict the equilibrium distribution of molecular systems. Inspired by the annealing process in thermodynamics, DiG uses deep neural networks to transform a simple distribution towards the equilibrium distribution, conditioned on a descriptor of a molecular system such as a chemical graph or a protein sequence. This framework enables the efficient generation of diverse conformations and provides estimations of state densities, orders of magnitude faster than conventional methods. We demonstrate applications of DiG on several molecular tasks, including protein conformation sampling, ligand structure sampling, catalyst–adsorbate sampling and property-guided structure generation. DiG presents a substantial advancement in methodology for statistically understanding molecular systems, opening up new research opportunities in the molecular sciences. Methods for predicting molecular structure predictions have so far focused on only the most probable conformation, but molecular structures are dynamic and can change when performing their biological functions, for example. Zheng et al. use a graph transformer approach to learn the equilibrium distribution of molecular systems and show that this can be helpful for a number of downstream tasks, including protein structure prediction, ligand docking and molecular design.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Nature Machine Intelligence
Multiple-
CiteScore
36.90
自引率
2.10%
发文量
127
期刊介绍:
Nature Machine Intelligence is a distinguished publication that presents original research and reviews on various topics in machine learning, robotics, and AI. Our focus extends beyond these fields, exploring their profound impact on other scientific disciplines, as well as societal and industrial aspects. We recognize limitless possibilities wherein machine intelligence can augment human capabilities and knowledge in domains like scientific exploration, healthcare, medical diagnostics, and the creation of safe and sustainable cities, transportation, and agriculture. Simultaneously, we acknowledge the emergence of ethical, social, and legal concerns due to the rapid pace of advancements.
To foster interdisciplinary discussions on these far-reaching implications, Nature Machine Intelligence serves as a platform for dialogue facilitated through Comments, News Features, News & Views articles, and Correspondence. Our goal is to encourage a comprehensive examination of these subjects.
Similar to all Nature-branded journals, Nature Machine Intelligence operates under the guidance of a team of skilled editors. We adhere to a fair and rigorous peer-review process, ensuring high standards of copy-editing and production, swift publication, and editorial independence.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


