Kemal Atalar, Yannic Rath, Rachel Crespo-Otero and George H. Booth
{"title":"通过相关电子波函数的变分插值实现快速准确的非绝热分子动力学","authors":"Kemal Atalar, Yannic Rath, Rachel Crespo-Otero and George H. Booth","doi":"10.1039/D4FD00062E","DOIUrl":null,"url":null,"abstract":"<p >We build on the concept of eigenvector continuation to develop an efficient multi-state method for the rigorous and smooth interpolation of a small training set of many-body wavefunctions through chemical space at mean-field cost. The inferred states are represented as variationally optimal linear combinations of the training states transferred between the many-body bases of different nuclear geometries. We show that analytic multi-state forces and nonadiabatic couplings from the model enable application to nonadiabatic molecular dynamics, developing an active learning scheme to ensure a compact and systematically improvable training set. This culminates in application to the nonadiabatic molecular dynamics of a photoexcited 28-atom hydrogen chain, with surprising complexity in the resulting nuclear motion. With just 22 DMRG calculations of training states from the low-energy correlated electronic structure at different geometries, we infer the multi-state energies, forces and nonadiabatic coupling vectors at 12 000 geometries with provable convergence to high accuracy along an ensemble of molecular trajectories, which would not be feasible with a brute force approach. This opens up a route to bridge the timescales between accurate single-point correlated electronic structure methods and timescales of relevance for photo-induced molecular dynamics.</p>","PeriodicalId":49075,"journal":{"name":"Faraday Discussions","volume":"254 ","pages":" 542-569"},"PeriodicalIF":3.1000,"publicationDate":"2024-05-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2024/fd/d4fd00062e?page=search","citationCount":"0","resultStr":"{\"title\":\"Fast and accurate nonadiabatic molecular dynamics enabled through variational interpolation of correlated electron wavefunctions†\",\"authors\":\"Kemal Atalar, Yannic Rath, Rachel Crespo-Otero and George H. Booth\",\"doi\":\"10.1039/D4FD00062E\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >We build on the concept of eigenvector continuation to develop an efficient multi-state method for the rigorous and smooth interpolation of a small training set of many-body wavefunctions through chemical space at mean-field cost. The inferred states are represented as variationally optimal linear combinations of the training states transferred between the many-body bases of different nuclear geometries. We show that analytic multi-state forces and nonadiabatic couplings from the model enable application to nonadiabatic molecular dynamics, developing an active learning scheme to ensure a compact and systematically improvable training set. This culminates in application to the nonadiabatic molecular dynamics of a photoexcited 28-atom hydrogen chain, with surprising complexity in the resulting nuclear motion. With just 22 DMRG calculations of training states from the low-energy correlated electronic structure at different geometries, we infer the multi-state energies, forces and nonadiabatic coupling vectors at 12 000 geometries with provable convergence to high accuracy along an ensemble of molecular trajectories, which would not be feasible with a brute force approach. This opens up a route to bridge the timescales between accurate single-point correlated electronic structure methods and timescales of relevance for photo-induced molecular dynamics.</p>\",\"PeriodicalId\":49075,\"journal\":{\"name\":\"Faraday Discussions\",\"volume\":\"254 \",\"pages\":\" 542-569\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2024-05-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.rsc.org/en/content/articlepdf/2024/fd/d4fd00062e?page=search\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Faraday Discussions\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2024/fd/d4fd00062e\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"Chemistry\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Faraday Discussions","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/fd/d4fd00062e","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Chemistry","Score":null,"Total":0}
Fast and accurate nonadiabatic molecular dynamics enabled through variational interpolation of correlated electron wavefunctions†
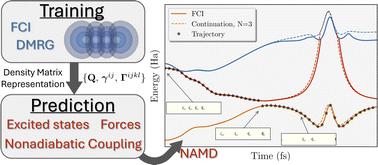
We build on the concept of eigenvector continuation to develop an efficient multi-state method for the rigorous and smooth interpolation of a small training set of many-body wavefunctions through chemical space at mean-field cost. The inferred states are represented as variationally optimal linear combinations of the training states transferred between the many-body bases of different nuclear geometries. We show that analytic multi-state forces and nonadiabatic couplings from the model enable application to nonadiabatic molecular dynamics, developing an active learning scheme to ensure a compact and systematically improvable training set. This culminates in application to the nonadiabatic molecular dynamics of a photoexcited 28-atom hydrogen chain, with surprising complexity in the resulting nuclear motion. With just 22 DMRG calculations of training states from the low-energy correlated electronic structure at different geometries, we infer the multi-state energies, forces and nonadiabatic coupling vectors at 12 000 geometries with provable convergence to high accuracy along an ensemble of molecular trajectories, which would not be feasible with a brute force approach. This opens up a route to bridge the timescales between accurate single-point correlated electronic structure methods and timescales of relevance for photo-induced molecular dynamics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


