Pabitra Narayan Samanta, Devashis Majumdar, Jerzy Leszczynski
{"title":"石墨烯和热塑性弹性体对定制沥青块体特性的影响:经典和量子模拟探索","authors":"Pabitra Narayan Samanta, Devashis Majumdar, Jerzy Leszczynski","doi":"10.1007/s11224-024-02330-4","DOIUrl":null,"url":null,"abstract":"<div><p>The modulation of bulk properties including the cohesive strength and the solubility of the asphaltenes, due to the inclusion of graphene nanosheets and the thermoplastic polymer, is probed by performing all-atom classical molecular dynamics (MD) simulations. The impact of morphological heterogeneity, including the size of the aromatic core of the asphaltene molecule, the nature of the heteroatom attached to the aromatic core, the orientation of the graphene nanosheets, and the surface area of the nanomaterial, on the bulk properties of the model systems of nanocomposites and interfaces is explored. The cohesive strength of the asphaltene composites is significantly enhanced by the introduction of graphene nanosheets. The addition of styrene–butadiene–styrene (SBS) block copolymer into the graphene-reinforced asphaltene systems improves the cohesive strength, structural plasticity, and compatibility between the nanomaterial and the asphaltenes. The <i>π</i>–<i>π</i> stacking interaction between the graphitic surface and the aromatic core of the asphaltene is identified to be the major driving force for modulating the cohesive strength. The dispersion interaction maximizes in the hierarchical layered structure compared to the randomly oriented structure of the graphene nanosheets and the asphaltene molecules. The energetics of non-covalent interaction are further assessed within the framework of dispersion-corrected density functional theory (DFT)-based methods. The DFT-derived adsorption energies and thermochemical properties substantiate the stronger interaction and the thermodynamic favorability of the adsorption processes in both the gas phase and solvent medium (toluene). The simulated IR and Raman spectra are also analyzed to reveal the nature of the interaction.</p></div>","PeriodicalId":780,"journal":{"name":"Structural Chemistry","volume":"35 4","pages":"1059 - 1073"},"PeriodicalIF":2.1000,"publicationDate":"2024-04-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Effects of graphene and thermoplastic elastomer on tailoring the bulk properties of asphaltenes: an exploration from classical and quantum simulations\",\"authors\":\"Pabitra Narayan Samanta, Devashis Majumdar, Jerzy Leszczynski\",\"doi\":\"10.1007/s11224-024-02330-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>The modulation of bulk properties including the cohesive strength and the solubility of the asphaltenes, due to the inclusion of graphene nanosheets and the thermoplastic polymer, is probed by performing all-atom classical molecular dynamics (MD) simulations. The impact of morphological heterogeneity, including the size of the aromatic core of the asphaltene molecule, the nature of the heteroatom attached to the aromatic core, the orientation of the graphene nanosheets, and the surface area of the nanomaterial, on the bulk properties of the model systems of nanocomposites and interfaces is explored. The cohesive strength of the asphaltene composites is significantly enhanced by the introduction of graphene nanosheets. The addition of styrene–butadiene–styrene (SBS) block copolymer into the graphene-reinforced asphaltene systems improves the cohesive strength, structural plasticity, and compatibility between the nanomaterial and the asphaltenes. The <i>π</i>–<i>π</i> stacking interaction between the graphitic surface and the aromatic core of the asphaltene is identified to be the major driving force for modulating the cohesive strength. The dispersion interaction maximizes in the hierarchical layered structure compared to the randomly oriented structure of the graphene nanosheets and the asphaltene molecules. The energetics of non-covalent interaction are further assessed within the framework of dispersion-corrected density functional theory (DFT)-based methods. The DFT-derived adsorption energies and thermochemical properties substantiate the stronger interaction and the thermodynamic favorability of the adsorption processes in both the gas phase and solvent medium (toluene). The simulated IR and Raman spectra are also analyzed to reveal the nature of the interaction.</p></div>\",\"PeriodicalId\":780,\"journal\":{\"name\":\"Structural Chemistry\",\"volume\":\"35 4\",\"pages\":\"1059 - 1073\"},\"PeriodicalIF\":2.1000,\"publicationDate\":\"2024-04-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Structural Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s11224-024-02330-4\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Structural Chemistry","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s11224-024-02330-4","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Effects of graphene and thermoplastic elastomer on tailoring the bulk properties of asphaltenes: an exploration from classical and quantum simulations
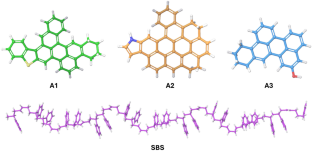
The modulation of bulk properties including the cohesive strength and the solubility of the asphaltenes, due to the inclusion of graphene nanosheets and the thermoplastic polymer, is probed by performing all-atom classical molecular dynamics (MD) simulations. The impact of morphological heterogeneity, including the size of the aromatic core of the asphaltene molecule, the nature of the heteroatom attached to the aromatic core, the orientation of the graphene nanosheets, and the surface area of the nanomaterial, on the bulk properties of the model systems of nanocomposites and interfaces is explored. The cohesive strength of the asphaltene composites is significantly enhanced by the introduction of graphene nanosheets. The addition of styrene–butadiene–styrene (SBS) block copolymer into the graphene-reinforced asphaltene systems improves the cohesive strength, structural plasticity, and compatibility between the nanomaterial and the asphaltenes. The π–π stacking interaction between the graphitic surface and the aromatic core of the asphaltene is identified to be the major driving force for modulating the cohesive strength. The dispersion interaction maximizes in the hierarchical layered structure compared to the randomly oriented structure of the graphene nanosheets and the asphaltene molecules. The energetics of non-covalent interaction are further assessed within the framework of dispersion-corrected density functional theory (DFT)-based methods. The DFT-derived adsorption energies and thermochemical properties substantiate the stronger interaction and the thermodynamic favorability of the adsorption processes in both the gas phase and solvent medium (toluene). The simulated IR and Raman spectra are also analyzed to reveal the nature of the interaction.
期刊介绍:
Structural Chemistry is an international forum for the publication of peer-reviewed original research papers that cover the condensed and gaseous states of matter and involve numerous techniques for the determination of structure and energetics, their results, and the conclusions derived from these studies. The journal overcomes the unnatural separation in the current literature among the areas of structure determination, energetics, and applications, as well as builds a bridge to other chemical disciplines. Ist comprehensive coverage encompasses broad discussion of results, observation of relationships among various properties, and the description and application of structure and energy information in all domains of chemistry.
We welcome the broadest range of accounts of research in structural chemistry involving the discussion of methodologies and structures,experimental, theoretical, and computational, and their combinations. We encourage discussions of structural information collected for their chemicaland biological significance.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


