Ramesh Boinpally, Lisa Borbridge, Veronica Wangsadipura
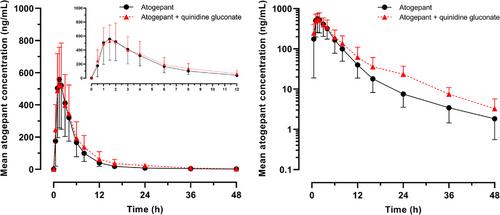
{"title":"健康参与者服用阿托格潘与葡萄糖酸奎尼丁的药代动力学和安全性:一期开放标签药物相互作用研究","authors":"Ramesh Boinpally, Lisa Borbridge, Veronica Wangsadipura","doi":"10.1002/cpdd.1407","DOIUrl":null,"url":null,"abstract":"<p>Atogepant, an oral calcitonin gene-related peptide receptor antagonist, is approved for the preventive treatment of migraine. Atogepant is a substrate of P-glycoprotein (P-gp), breast cancer resistance protein, organic anion transporting polypeptide transporters, and cytochrome P450 (CYP)3A4 and 2D6. Quinidine is a strong P-gp and CYP2D6 inhibitor. A phase 1 open-label study evaluated the effect of P-gp and CYP2D6 inhibition by quinidine on the pharmacokinetics of atogepant, and the safety and tolerability of atogepant and quinidine gluconate (QG) when co-administered and when given alone in 33 healthy adults. There was no significant change in the atogepant maximum plasma concentration with QG co-administration. The overall systemic exposure, the area under the plasma concentration-time curve (from time 0 to time t or to infinity), of atogepant increased by 25% when co-administered with QG. However, such an increase was not considered clinically relevant. Atogepant did not alter the mean plasma concentration of quinidine at steady state. The incidence of treatment-emergent adverse events (TEAEs) was highest when QG was administered alone (42.4%), which was primarily due to QT prolongation. Most TEAEs reported were mild in severity and resolved within 1-2 days. Co-administration of atogepant with QG did not result in any unexpected tolerability findings in this phase 1 study in healthy participants. The increase in atogepant exposure during QG co-administration could be due to inhibition of CYP2D6 (a minor contributor to atogepant clearance) as well as inhibition of P-gp.</p>","PeriodicalId":10495,"journal":{"name":"Clinical Pharmacology in Drug Development","volume":"13 8","pages":"930-937"},"PeriodicalIF":1.5000,"publicationDate":"2024-05-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/cpdd.1407","citationCount":"0","resultStr":"{\"title\":\"Pharmacokinetics and Safety of Atogepant Co-administered with Quinidine Gluconate in Healthy Participants: A Phase 1, Open-Label, Drug-Drug Interaction Study\",\"authors\":\"Ramesh Boinpally, Lisa Borbridge, Veronica Wangsadipura\",\"doi\":\"10.1002/cpdd.1407\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Atogepant, an oral calcitonin gene-related peptide receptor antagonist, is approved for the preventive treatment of migraine. Atogepant is a substrate of P-glycoprotein (P-gp), breast cancer resistance protein, organic anion transporting polypeptide transporters, and cytochrome P450 (CYP)3A4 and 2D6. Quinidine is a strong P-gp and CYP2D6 inhibitor. A phase 1 open-label study evaluated the effect of P-gp and CYP2D6 inhibition by quinidine on the pharmacokinetics of atogepant, and the safety and tolerability of atogepant and quinidine gluconate (QG) when co-administered and when given alone in 33 healthy adults. There was no significant change in the atogepant maximum plasma concentration with QG co-administration. The overall systemic exposure, the area under the plasma concentration-time curve (from time 0 to time t or to infinity), of atogepant increased by 25% when co-administered with QG. However, such an increase was not considered clinically relevant. Atogepant did not alter the mean plasma concentration of quinidine at steady state. The incidence of treatment-emergent adverse events (TEAEs) was highest when QG was administered alone (42.4%), which was primarily due to QT prolongation. Most TEAEs reported were mild in severity and resolved within 1-2 days. Co-administration of atogepant with QG did not result in any unexpected tolerability findings in this phase 1 study in healthy participants. The increase in atogepant exposure during QG co-administration could be due to inhibition of CYP2D6 (a minor contributor to atogepant clearance) as well as inhibition of P-gp.</p>\",\"PeriodicalId\":10495,\"journal\":{\"name\":\"Clinical Pharmacology in Drug Development\",\"volume\":\"13 8\",\"pages\":\"930-937\"},\"PeriodicalIF\":1.5000,\"publicationDate\":\"2024-05-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/cpdd.1407\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical Pharmacology in Drug Development\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/cpdd.1407\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"PHARMACOLOGY & PHARMACY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Pharmacology in Drug Development","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/cpdd.1407","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"PHARMACOLOGY & PHARMACY","Score":null,"Total":0}
Pharmacokinetics and Safety of Atogepant Co-administered with Quinidine Gluconate in Healthy Participants: A Phase 1, Open-Label, Drug-Drug Interaction Study
Atogepant, an oral calcitonin gene-related peptide receptor antagonist, is approved for the preventive treatment of migraine. Atogepant is a substrate of P-glycoprotein (P-gp), breast cancer resistance protein, organic anion transporting polypeptide transporters, and cytochrome P450 (CYP)3A4 and 2D6. Quinidine is a strong P-gp and CYP2D6 inhibitor. A phase 1 open-label study evaluated the effect of P-gp and CYP2D6 inhibition by quinidine on the pharmacokinetics of atogepant, and the safety and tolerability of atogepant and quinidine gluconate (QG) when co-administered and when given alone in 33 healthy adults. There was no significant change in the atogepant maximum plasma concentration with QG co-administration. The overall systemic exposure, the area under the plasma concentration-time curve (from time 0 to time t or to infinity), of atogepant increased by 25% when co-administered with QG. However, such an increase was not considered clinically relevant. Atogepant did not alter the mean plasma concentration of quinidine at steady state. The incidence of treatment-emergent adverse events (TEAEs) was highest when QG was administered alone (42.4%), which was primarily due to QT prolongation. Most TEAEs reported were mild in severity and resolved within 1-2 days. Co-administration of atogepant with QG did not result in any unexpected tolerability findings in this phase 1 study in healthy participants. The increase in atogepant exposure during QG co-administration could be due to inhibition of CYP2D6 (a minor contributor to atogepant clearance) as well as inhibition of P-gp.
期刊介绍:
Clinical Pharmacology in Drug Development is an international, peer-reviewed, online publication focused on publishing high-quality clinical pharmacology studies in drug development which are primarily (but not exclusively) performed in early development phases in healthy subjects.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


