Şevval Aktürk, Igor Mapelli, Merve N. Güler, Kanat Gürün, Büşra Katırcıoğlu, Kıvılcım Başak Vural, Ekin Sağlıcan, Mehmet Çetin, Reyhan Yaka, Elif Sürer, Gözde Atağ, Sevim Seda Çokoğlu, Arda Sevkar, N. Ezgi Altınışık, Dilek Koptekin, Mehmet Somel
{"title":"利用血统模拟为古代基因组的亲缘关系估算工具设定基准","authors":"Şevval Aktürk, Igor Mapelli, Merve N. Güler, Kanat Gürün, Büşra Katırcıoğlu, Kıvılcım Başak Vural, Ekin Sağlıcan, Mehmet Çetin, Reyhan Yaka, Elif Sürer, Gözde Atağ, Sevim Seda Çokoğlu, Arda Sevkar, N. Ezgi Altınışık, Dilek Koptekin, Mehmet Somel","doi":"10.1111/1755-0998.13960","DOIUrl":null,"url":null,"abstract":"<p>There is growing interest in uncovering genetic kinship patterns in past societies using low-coverage palaeogenomes. Here, we benchmark four tools for kinship estimation with such data: lcMLkin, NgsRelate, KIN, and READ, which differ in their input, IBD estimation methods, and statistical approaches. We used pedigree and ancient genome sequence simulations to evaluate these tools when only a limited number (1 to 50 K, with minor allele frequency ≥0.01) of shared SNPs are available. The performance of all four tools was comparable using ≥20 K SNPs. We found that first-degree related pairs can be accurately classified even with 1 K SNPs, with 85% <i>F</i><sub>1</sub> scores using READ and 96% using NgsRelate or lcMLkin. Distinguishing third-degree relatives from unrelated pairs or second-degree relatives was also possible with high accuracy (<i>F</i><sub>1</sub> > 90%) with 5 K SNPs using NgsRelate and lcMLkin, while READ and KIN showed lower success (69 and 79% respectively). Meanwhile, noise in population allele frequencies and inbreeding (first-cousin mating) led to deviations in kinship coefficients, with different sensitivities across tools. We conclude that using multiple tools in parallel might be an effective approach to achieve robust estimates on ultra-low-coverage genomes.</p>","PeriodicalId":211,"journal":{"name":"Molecular Ecology Resources","volume":"24 5","pages":""},"PeriodicalIF":5.5000,"publicationDate":"2024-04-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/1755-0998.13960","citationCount":"0","resultStr":"{\"title\":\"Benchmarking kinship estimation tools for ancient genomes using pedigree simulations\",\"authors\":\"Şevval Aktürk, Igor Mapelli, Merve N. Güler, Kanat Gürün, Büşra Katırcıoğlu, Kıvılcım Başak Vural, Ekin Sağlıcan, Mehmet Çetin, Reyhan Yaka, Elif Sürer, Gözde Atağ, Sevim Seda Çokoğlu, Arda Sevkar, N. Ezgi Altınışık, Dilek Koptekin, Mehmet Somel\",\"doi\":\"10.1111/1755-0998.13960\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>There is growing interest in uncovering genetic kinship patterns in past societies using low-coverage palaeogenomes. Here, we benchmark four tools for kinship estimation with such data: lcMLkin, NgsRelate, KIN, and READ, which differ in their input, IBD estimation methods, and statistical approaches. We used pedigree and ancient genome sequence simulations to evaluate these tools when only a limited number (1 to 50 K, with minor allele frequency ≥0.01) of shared SNPs are available. The performance of all four tools was comparable using ≥20 K SNPs. We found that first-degree related pairs can be accurately classified even with 1 K SNPs, with 85% <i>F</i><sub>1</sub> scores using READ and 96% using NgsRelate or lcMLkin. Distinguishing third-degree relatives from unrelated pairs or second-degree relatives was also possible with high accuracy (<i>F</i><sub>1</sub> > 90%) with 5 K SNPs using NgsRelate and lcMLkin, while READ and KIN showed lower success (69 and 79% respectively). Meanwhile, noise in population allele frequencies and inbreeding (first-cousin mating) led to deviations in kinship coefficients, with different sensitivities across tools. We conclude that using multiple tools in parallel might be an effective approach to achieve robust estimates on ultra-low-coverage genomes.</p>\",\"PeriodicalId\":211,\"journal\":{\"name\":\"Molecular Ecology Resources\",\"volume\":\"24 5\",\"pages\":\"\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2024-04-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1111/1755-0998.13960\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Ecology Resources\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/1755-0998.13960\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Ecology Resources","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/1755-0998.13960","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
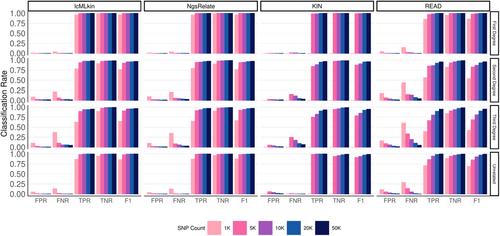
人们对利用低覆盖率古基因组揭示过去社会的遗传亲缘关系模式越来越感兴趣。在此,我们对使用此类数据进行亲缘关系估计的四种工具进行了基准测试:lcMLkin、NgsRelate、KIN 和 READ,它们在输入、IBD 估计方法和统计方法上各不相同。我们使用血统和古基因组序列模拟来评估这些工具,当只有有限数量(1 到 50 K,小等位基因频率≥0.01)的共享 SNP 可用时。使用≥20 K SNPs时,所有四种工具的性能相当。我们发现,即使只有 1 K 个 SNPs,也能对一级亲属配对进行准确分类,使用 READ 的 F1 得分率为 85%,使用 NgsRelate 或 lcMLkin 的 F1 得分率为 96%。利用 5 K SNPs,使用 NgsRelate 和 lcMLkin 也能以较高的准确率(F1 > 90%)将三代亲属与无亲属关系的配对或二代亲属区分开来,而 READ 和 KIN 的成功率较低(分别为 69% 和 79%)。同时,种群等位基因频率和近亲繁殖(嫡亲交配)的噪声导致亲缘关系系数出现偏差,不同工具的敏感度也不同。我们的结论是,并行使用多种工具可能是在超低覆盖率基因组上实现稳健估计的有效方法。
Benchmarking kinship estimation tools for ancient genomes using pedigree simulations
There is growing interest in uncovering genetic kinship patterns in past societies using low-coverage palaeogenomes. Here, we benchmark four tools for kinship estimation with such data: lcMLkin, NgsRelate, KIN, and READ, which differ in their input, IBD estimation methods, and statistical approaches. We used pedigree and ancient genome sequence simulations to evaluate these tools when only a limited number (1 to 50 K, with minor allele frequency ≥0.01) of shared SNPs are available. The performance of all four tools was comparable using ≥20 K SNPs. We found that first-degree related pairs can be accurately classified even with 1 K SNPs, with 85% F1 scores using READ and 96% using NgsRelate or lcMLkin. Distinguishing third-degree relatives from unrelated pairs or second-degree relatives was also possible with high accuracy (F1 > 90%) with 5 K SNPs using NgsRelate and lcMLkin, while READ and KIN showed lower success (69 and 79% respectively). Meanwhile, noise in population allele frequencies and inbreeding (first-cousin mating) led to deviations in kinship coefficients, with different sensitivities across tools. We conclude that using multiple tools in parallel might be an effective approach to achieve robust estimates on ultra-low-coverage genomes.
期刊介绍:
Molecular Ecology Resources promotes the creation of comprehensive resources for the scientific community, encompassing computer programs, statistical and molecular advancements, and a diverse array of molecular tools. Serving as a conduit for disseminating these resources, the journal targets a broad audience of researchers in the fields of evolution, ecology, and conservation. Articles in Molecular Ecology Resources are crafted to support investigations tackling significant questions within these disciplines.
In addition to original resource articles, Molecular Ecology Resources features Reviews, Opinions, and Comments relevant to the field. The journal also periodically releases Special Issues focusing on resource development within specific areas.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


