Inez Bosnyak, Mustafa Sadek, Wasantha Ranatunga, Tamas Kozicz, Eva Morava
{"title":"转铁蛋白糖基化正常并不能排除严重的 ALG1 缺乏症","authors":"Inez Bosnyak, Mustafa Sadek, Wasantha Ranatunga, Tamas Kozicz, Eva Morava","doi":"10.1002/jmd2.12415","DOIUrl":null,"url":null,"abstract":"<p>ALG1-CDG is a rare, clinically variable metabolic disease, caused by the defect of adding the first mannose (Man) to N-acetylglucosamine (GlcNAc<sub>2</sub>)-pyrophosphate (PP)-dolichol to the growing oligosaccharide chain, resulting in impaired N-glycosylation of proteins. N-glycosylation has a key role in functionality, stability, and half-life of most proteins. Therefore, congenital defects of glycosylation typically are multisystem disorders. Here we report a 3-year-old patient with severe neurological, cardiovascular, respiratory, musculoskeletal and gastrointestinal symptoms. ALG1-CDG was suggested based on exome sequencing and Western blot analysis. Despite her severe clinical manifestations and genetic diagnosis, serum transferrin glycoform analysis was normal. Western blot analysis of highly glycosylated proteins in fibroblasts revealed decreased intercellular adhesion molecule 1 (ICAM1), but normal lysosomal associated membrane protein 1 and 2 (LAMP1 and LAMP2) expression levels. Glycoproteomics in fibroblasts showed the presence of the abnormal tetrasacharide. Reviewing the literature, we found 86 reported ALG1-CDG patients, but only one with normal transferrin analysis. Based on our results we would like to highlight the importance of multiple approaches in diagnosing ALG1-CDG, as normal serum transferrin glycosylation or other biomarkers with normal expression levels can occur.</p>","PeriodicalId":14930,"journal":{"name":"JIMD reports","volume":"65 3","pages":"135-143"},"PeriodicalIF":1.8000,"publicationDate":"2024-04-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jmd2.12415","citationCount":"0","resultStr":"{\"title\":\"Normal transferrin glycosylation does not rule out severe ALG1 deficiency\",\"authors\":\"Inez Bosnyak, Mustafa Sadek, Wasantha Ranatunga, Tamas Kozicz, Eva Morava\",\"doi\":\"10.1002/jmd2.12415\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>ALG1-CDG is a rare, clinically variable metabolic disease, caused by the defect of adding the first mannose (Man) to N-acetylglucosamine (GlcNAc<sub>2</sub>)-pyrophosphate (PP)-dolichol to the growing oligosaccharide chain, resulting in impaired N-glycosylation of proteins. N-glycosylation has a key role in functionality, stability, and half-life of most proteins. Therefore, congenital defects of glycosylation typically are multisystem disorders. Here we report a 3-year-old patient with severe neurological, cardiovascular, respiratory, musculoskeletal and gastrointestinal symptoms. ALG1-CDG was suggested based on exome sequencing and Western blot analysis. Despite her severe clinical manifestations and genetic diagnosis, serum transferrin glycoform analysis was normal. Western blot analysis of highly glycosylated proteins in fibroblasts revealed decreased intercellular adhesion molecule 1 (ICAM1), but normal lysosomal associated membrane protein 1 and 2 (LAMP1 and LAMP2) expression levels. Glycoproteomics in fibroblasts showed the presence of the abnormal tetrasacharide. Reviewing the literature, we found 86 reported ALG1-CDG patients, but only one with normal transferrin analysis. Based on our results we would like to highlight the importance of multiple approaches in diagnosing ALG1-CDG, as normal serum transferrin glycosylation or other biomarkers with normal expression levels can occur.</p>\",\"PeriodicalId\":14930,\"journal\":{\"name\":\"JIMD reports\",\"volume\":\"65 3\",\"pages\":\"135-143\"},\"PeriodicalIF\":1.8000,\"publicationDate\":\"2024-04-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jmd2.12415\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"JIMD reports\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jmd2.12415\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"JIMD reports","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jmd2.12415","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
Normal transferrin glycosylation does not rule out severe ALG1 deficiency
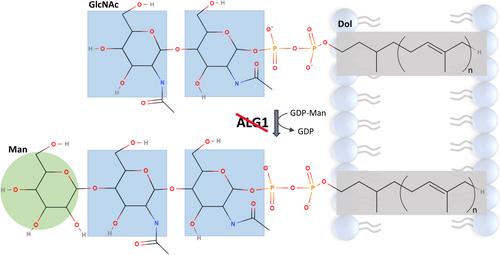
ALG1-CDG is a rare, clinically variable metabolic disease, caused by the defect of adding the first mannose (Man) to N-acetylglucosamine (GlcNAc2)-pyrophosphate (PP)-dolichol to the growing oligosaccharide chain, resulting in impaired N-glycosylation of proteins. N-glycosylation has a key role in functionality, stability, and half-life of most proteins. Therefore, congenital defects of glycosylation typically are multisystem disorders. Here we report a 3-year-old patient with severe neurological, cardiovascular, respiratory, musculoskeletal and gastrointestinal symptoms. ALG1-CDG was suggested based on exome sequencing and Western blot analysis. Despite her severe clinical manifestations and genetic diagnosis, serum transferrin glycoform analysis was normal. Western blot analysis of highly glycosylated proteins in fibroblasts revealed decreased intercellular adhesion molecule 1 (ICAM1), but normal lysosomal associated membrane protein 1 and 2 (LAMP1 and LAMP2) expression levels. Glycoproteomics in fibroblasts showed the presence of the abnormal tetrasacharide. Reviewing the literature, we found 86 reported ALG1-CDG patients, but only one with normal transferrin analysis. Based on our results we would like to highlight the importance of multiple approaches in diagnosing ALG1-CDG, as normal serum transferrin glycosylation or other biomarkers with normal expression levels can occur.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


