Koichi Handa*, Saki Yoshimura, Michiharu Kageyama and Takeshi Iijima,
{"title":"通过机器反复学习开发 QSAR 建模的新方法:药物在各组织分布的案例研究。","authors":"Koichi Handa*, Saki Yoshimura, Michiharu Kageyama and Takeshi Iijima, ","doi":"10.1021/acs.jcim.4c00046","DOIUrl":null,"url":null,"abstract":"<p >Artificial intelligence is expected to help identify excellent candidates in drug discovery. However, we face a lack of data, as it is time-consuming and expensive to acquire raw data perfectly for many compounds. Hence, we tried to develop a novel quantitative structure-activity relationship (QSAR) method to predict a parameter more precisely from an incomplete data set via optimizing data handling by making use of predicted explanatory variables. As a case study we focused on the tissue-to-plasma partition coefficient (Kp), which is an important parameter for understanding drug distribution in tissues and building the physiologically based pharmacokinetic model and is a representative of small and sparse data sets. In this study, we predicted the Kp values of 119 compounds in nine tissues (adipose, brain, gut, heart, kidney, liver, lung, muscle, and skin), although some of these were not available. To fill the missing values in Kp for each tissue, first we predicted those Kp values by the nonmissing data set using a random forest (RF) model with <i>in vitro</i> parameters (log P, fu, Drug Class, and fi) like a classical prediction by a QSAR model. Next, to predict the tissue-specific Kp values in a test data set, we constructed a second RF model with not only <i>in vitro</i> parameters but also the Kp values of other tissues (i.e., other than target tissues) predicted by the first RF model as explanatory variables. Furthermore, we tested all possible combinations of explanatory variables and selected the model with the highest predictability from the test data set as the final model. The evaluation of Kp prediction accuracy based on the root-mean-square error and <i>R</i><sup>2</sup> value revealed that the proposed models outperformed other machine learning methods such as the conventional RF and message-passing neural networks. Significant improvements were observed in the Kp values of adipose tissue, brain, kidney, liver, and skin. These improvements indicated that the Kp information on other tissues can be used to predict the same for a specific tissue. Additionally, we found a novel relationship between each tissue by evaluating all combinations of explanatory variables. In conclusion, we developed a novel RF model to predict Kp values. We hope that this method will be applied to various problems in the field of experimental biology which often contains missing values in the near future.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":"64 9","pages":"3662–3669"},"PeriodicalIF":5.3000,"publicationDate":"2024-04-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Development of Novel Methods for QSAR Modeling by Machine Learning Repeatedly: A Case Study on Drug Distribution to Each Tissue\",\"authors\":\"Koichi Handa*, Saki Yoshimura, Michiharu Kageyama and Takeshi Iijima, \",\"doi\":\"10.1021/acs.jcim.4c00046\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Artificial intelligence is expected to help identify excellent candidates in drug discovery. However, we face a lack of data, as it is time-consuming and expensive to acquire raw data perfectly for many compounds. Hence, we tried to develop a novel quantitative structure-activity relationship (QSAR) method to predict a parameter more precisely from an incomplete data set via optimizing data handling by making use of predicted explanatory variables. As a case study we focused on the tissue-to-plasma partition coefficient (Kp), which is an important parameter for understanding drug distribution in tissues and building the physiologically based pharmacokinetic model and is a representative of small and sparse data sets. In this study, we predicted the Kp values of 119 compounds in nine tissues (adipose, brain, gut, heart, kidney, liver, lung, muscle, and skin), although some of these were not available. To fill the missing values in Kp for each tissue, first we predicted those Kp values by the nonmissing data set using a random forest (RF) model with <i>in vitro</i> parameters (log P, fu, Drug Class, and fi) like a classical prediction by a QSAR model. Next, to predict the tissue-specific Kp values in a test data set, we constructed a second RF model with not only <i>in vitro</i> parameters but also the Kp values of other tissues (i.e., other than target tissues) predicted by the first RF model as explanatory variables. Furthermore, we tested all possible combinations of explanatory variables and selected the model with the highest predictability from the test data set as the final model. The evaluation of Kp prediction accuracy based on the root-mean-square error and <i>R</i><sup>2</sup> value revealed that the proposed models outperformed other machine learning methods such as the conventional RF and message-passing neural networks. Significant improvements were observed in the Kp values of adipose tissue, brain, kidney, liver, and skin. These improvements indicated that the Kp information on other tissues can be used to predict the same for a specific tissue. Additionally, we found a novel relationship between each tissue by evaluating all combinations of explanatory variables. In conclusion, we developed a novel RF model to predict Kp values. We hope that this method will be applied to various problems in the field of experimental biology which often contains missing values in the near future.</p>\",\"PeriodicalId\":44,\"journal\":{\"name\":\"Journal of Chemical Information and Modeling \",\"volume\":\"64 9\",\"pages\":\"3662–3669\"},\"PeriodicalIF\":5.3000,\"publicationDate\":\"2024-04-19\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Information and Modeling \",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jcim.4c00046\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jcim.4c00046","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
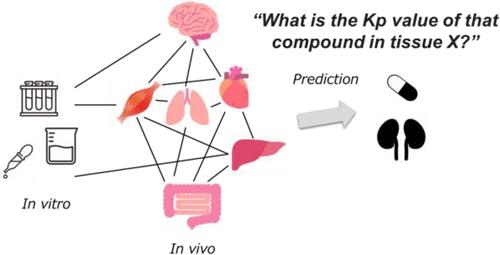
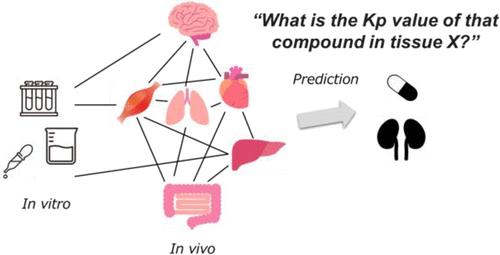
Development of Novel Methods for QSAR Modeling by Machine Learning Repeatedly: A Case Study on Drug Distribution to Each Tissue
Artificial intelligence is expected to help identify excellent candidates in drug discovery. However, we face a lack of data, as it is time-consuming and expensive to acquire raw data perfectly for many compounds. Hence, we tried to develop a novel quantitative structure-activity relationship (QSAR) method to predict a parameter more precisely from an incomplete data set via optimizing data handling by making use of predicted explanatory variables. As a case study we focused on the tissue-to-plasma partition coefficient (Kp), which is an important parameter for understanding drug distribution in tissues and building the physiologically based pharmacokinetic model and is a representative of small and sparse data sets. In this study, we predicted the Kp values of 119 compounds in nine tissues (adipose, brain, gut, heart, kidney, liver, lung, muscle, and skin), although some of these were not available. To fill the missing values in Kp for each tissue, first we predicted those Kp values by the nonmissing data set using a random forest (RF) model with in vitro parameters (log P, fu, Drug Class, and fi) like a classical prediction by a QSAR model. Next, to predict the tissue-specific Kp values in a test data set, we constructed a second RF model with not only in vitro parameters but also the Kp values of other tissues (i.e., other than target tissues) predicted by the first RF model as explanatory variables. Furthermore, we tested all possible combinations of explanatory variables and selected the model with the highest predictability from the test data set as the final model. The evaluation of Kp prediction accuracy based on the root-mean-square error and R2 value revealed that the proposed models outperformed other machine learning methods such as the conventional RF and message-passing neural networks. Significant improvements were observed in the Kp values of adipose tissue, brain, kidney, liver, and skin. These improvements indicated that the Kp information on other tissues can be used to predict the same for a specific tissue. Additionally, we found a novel relationship between each tissue by evaluating all combinations of explanatory variables. In conclusion, we developed a novel RF model to predict Kp values. We hope that this method will be applied to various problems in the field of experimental biology which often contains missing values in the near future.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


