Manu Juneja, Andrew Wei, Kylie Mason, Tamia Nguyen, John F Seymour, Surender Juneja
{"title":"伪装成伯基特淋巴瘤的 CD34-TdT-B-ALL","authors":"Manu Juneja, Andrew Wei, Kylie Mason, Tamia Nguyen, John F Seymour, Surender Juneja","doi":"10.1002/jha2.884","DOIUrl":null,"url":null,"abstract":"<p>A 23-year-old female presented with widespread petechiae and oral mucosal bleeding. There was no palpable lymphadenopathy or hepatosplenomegaly. Complete blood count included haemoglobin 77 g/L (normal 115–155), white cell count 20.4 × 10<sup>9</sup>/L (4.0–12.0), “blasts” 10.6 (52%), neutrophils 3.3 × 10<sup>9</sup>/L (2.0–8.0), platelets 11 × 10<sup>9</sup>/L (150–400). Peripheral blood morphology demonstrated monomorphic immature mononuclear cells with deeply basophilic cytoplasm and numerous small cytoplasmic vacuoles (Figure 1, left image; 1000x magnification) and markedly elevated serum lactate dehydrogenase (14358 IU; 120–250), suggestive of Burkitt leukaemia/lymphoma. Blood was analysed using standard methods as outlined by the ICSH (International Committee for Standardization in Haematology). Immunophenotyping demonstrated CD10+, CD19+ CD20+, CD22+,CD24+, CD34-, CD38+, CD45+ (dim) and CD58+ with cytoplasmic lambda light chain restriction (without surface light chain expression) consistent with a B-cell lymphoma or pre-B ALL. Bone marrow was markedly hypercellular with 83% neoplastic cells with identical morphology (Figure 1, middle image; 1000x magnification). Immunohistochemistry was negative for TdT and CD34. Chromosomal studies identified an unbalanced t(5;8)(q13;p21) resulting in the loss of part of the long arm of chromosome 5, and deletion of 9p by microarray. Fluorescent in-situ hybridisation did not identify <i>MYC</i> rearrangements typical of Burkitt lymphoma. Targeted molecular studies identified <i>NRAS</i> and <i>BRAF</i> variants and whole genome and transcriptome analysis demonstrated a <i>MEF2D::HNRNPUL1</i> rearrangement (Figure 1, right image). Prior to molecular results being available, the patient was commenced on R-CODOX-M/R-IVAC, achieving complete remission. With full diagnostic information available, consolidation delivered an ALL regimen. This case highlights how, despite a thorough investigation by all appropriate modalities including morphology, immunophenotyping and cytogenetics, the final correct diagnosis may be delayed until the determination of the molecular profile. Further complicating this case, B-ALL with MEF2D has only recently been described under the category of B-lymphoblastic leukaemia/lymphoma with other defined genetic abnormalities (WHO, 2022).</p><p>The authors declare no conflict of interest.</p><p>Patient has moved overseas so consent is not feasible. Patient's details are\nadequately anonymised.</p><p>N/A</p><p>All patient treatment protocols in our institution are agreed to by the Institutional Ethics Committee.</p>","PeriodicalId":72883,"journal":{"name":"EJHaem","volume":null,"pages":null},"PeriodicalIF":0.0000,"publicationDate":"2024-04-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jha2.884","citationCount":"0","resultStr":"{\"title\":\"CD34-TdT-B-ALL masquerading as Burkitt lymphoma\",\"authors\":\"Manu Juneja, Andrew Wei, Kylie Mason, Tamia Nguyen, John F Seymour, Surender Juneja\",\"doi\":\"10.1002/jha2.884\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>A 23-year-old female presented with widespread petechiae and oral mucosal bleeding. There was no palpable lymphadenopathy or hepatosplenomegaly. Complete blood count included haemoglobin 77 g/L (normal 115–155), white cell count 20.4 × 10<sup>9</sup>/L (4.0–12.0), “blasts” 10.6 (52%), neutrophils 3.3 × 10<sup>9</sup>/L (2.0–8.0), platelets 11 × 10<sup>9</sup>/L (150–400). Peripheral blood morphology demonstrated monomorphic immature mononuclear cells with deeply basophilic cytoplasm and numerous small cytoplasmic vacuoles (Figure 1, left image; 1000x magnification) and markedly elevated serum lactate dehydrogenase (14358 IU; 120–250), suggestive of Burkitt leukaemia/lymphoma. Blood was analysed using standard methods as outlined by the ICSH (International Committee for Standardization in Haematology). Immunophenotyping demonstrated CD10+, CD19+ CD20+, CD22+,CD24+, CD34-, CD38+, CD45+ (dim) and CD58+ with cytoplasmic lambda light chain restriction (without surface light chain expression) consistent with a B-cell lymphoma or pre-B ALL. Bone marrow was markedly hypercellular with 83% neoplastic cells with identical morphology (Figure 1, middle image; 1000x magnification). Immunohistochemistry was negative for TdT and CD34. Chromosomal studies identified an unbalanced t(5;8)(q13;p21) resulting in the loss of part of the long arm of chromosome 5, and deletion of 9p by microarray. Fluorescent in-situ hybridisation did not identify <i>MYC</i> rearrangements typical of Burkitt lymphoma. Targeted molecular studies identified <i>NRAS</i> and <i>BRAF</i> variants and whole genome and transcriptome analysis demonstrated a <i>MEF2D::HNRNPUL1</i> rearrangement (Figure 1, right image). Prior to molecular results being available, the patient was commenced on R-CODOX-M/R-IVAC, achieving complete remission. With full diagnostic information available, consolidation delivered an ALL regimen. This case highlights how, despite a thorough investigation by all appropriate modalities including morphology, immunophenotyping and cytogenetics, the final correct diagnosis may be delayed until the determination of the molecular profile. Further complicating this case, B-ALL with MEF2D has only recently been described under the category of B-lymphoblastic leukaemia/lymphoma with other defined genetic abnormalities (WHO, 2022).</p><p>The authors declare no conflict of interest.</p><p>Patient has moved overseas so consent is not feasible. Patient's details are\\nadequately anonymised.</p><p>N/A</p><p>All patient treatment protocols in our institution are agreed to by the Institutional Ethics Committee.</p>\",\"PeriodicalId\":72883,\"journal\":{\"name\":\"EJHaem\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2024-04-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jha2.884\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"EJHaem\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jha2.884\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"EJHaem","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jha2.884","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
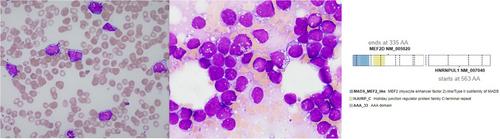
A 23-year-old female presented with widespread petechiae and oral mucosal bleeding. There was no palpable lymphadenopathy or hepatosplenomegaly. Complete blood count included haemoglobin 77 g/L (normal 115–155), white cell count 20.4 × 109/L (4.0–12.0), “blasts” 10.6 (52%), neutrophils 3.3 × 109/L (2.0–8.0), platelets 11 × 109/L (150–400). Peripheral blood morphology demonstrated monomorphic immature mononuclear cells with deeply basophilic cytoplasm and numerous small cytoplasmic vacuoles (Figure 1, left image; 1000x magnification) and markedly elevated serum lactate dehydrogenase (14358 IU; 120–250), suggestive of Burkitt leukaemia/lymphoma. Blood was analysed using standard methods as outlined by the ICSH (International Committee for Standardization in Haematology). Immunophenotyping demonstrated CD10+, CD19+ CD20+, CD22+,CD24+, CD34-, CD38+, CD45+ (dim) and CD58+ with cytoplasmic lambda light chain restriction (without surface light chain expression) consistent with a B-cell lymphoma or pre-B ALL. Bone marrow was markedly hypercellular with 83% neoplastic cells with identical morphology (Figure 1, middle image; 1000x magnification). Immunohistochemistry was negative for TdT and CD34. Chromosomal studies identified an unbalanced t(5;8)(q13;p21) resulting in the loss of part of the long arm of chromosome 5, and deletion of 9p by microarray. Fluorescent in-situ hybridisation did not identify MYC rearrangements typical of Burkitt lymphoma. Targeted molecular studies identified NRAS and BRAF variants and whole genome and transcriptome analysis demonstrated a MEF2D::HNRNPUL1 rearrangement (Figure 1, right image). Prior to molecular results being available, the patient was commenced on R-CODOX-M/R-IVAC, achieving complete remission. With full diagnostic information available, consolidation delivered an ALL regimen. This case highlights how, despite a thorough investigation by all appropriate modalities including morphology, immunophenotyping and cytogenetics, the final correct diagnosis may be delayed until the determination of the molecular profile. Further complicating this case, B-ALL with MEF2D has only recently been described under the category of B-lymphoblastic leukaemia/lymphoma with other defined genetic abnormalities (WHO, 2022).
The authors declare no conflict of interest.
Patient has moved overseas so consent is not feasible. Patient's details are
adequately anonymised.
N/A
All patient treatment protocols in our institution are agreed to by the Institutional Ethics Committee.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


