{"title":"碱土金属氧化物的化学储热机制:氧化镁(001)表面初始水化反应的 Ab Initio 模型","authors":"Toyokazu Ishida*, and , Kazuya Ishimura, ","doi":"10.1021/acs.jpcc.3c08408","DOIUrl":null,"url":null,"abstract":"<p >Effective management of reusable waste heat is one of the most important technologies for reducing energy consumption and promoting energy conservation. In particular, chemical heat storage is a practical solution to the spatial and temporal mismatches of energy use. Among various chemical reactions, the reversible hydration process of periclase (MgO) to brucite [Mg(OH)<sub>2</sub>] is a practical candidate for the future applications in various social systems. In this article, in order to clarify the atomistic reaction mechanism of thermochemical heat storage in the MgO hydration reaction, we calculated potential energy profiles to understand the complex thermochemical reaction on the solid surface. By assuming a series of elementary reaction processes in the early stage of the surface reaction, we have practically estimated the reaction energetics for the hydration reaction in both the MgO(001) terrace and step geometries. First, we calculate two important thermodynamic parameters that characterize the thermochemical reaction (the reaction heat and the transition temperature). Our computational results show good agreement with the available thermodynamic data. Then, to clarify the nature of the thermochemical heat storage process, we introduce the 2D potential energy diagram, which is determined by the dislocation distance of the Mg atoms and the coordination (solvation) number of the Mg atoms. This 2D potential energy diagram clearly shows that the activation energy is highly dependent on the surface geometry of MgO, while an atomistic process in this rate-determining step (an extraction of Mg atoms from the inner layer to the surface aqueous layer) is exactly the same in both the terrace and step surface. These computational results lead to a simple guideline for the design of thermochemical heat storage systems.</p>","PeriodicalId":61,"journal":{"name":"The Journal of Physical Chemistry C","volume":"128 18","pages":"7397–7407"},"PeriodicalIF":3.2000,"publicationDate":"2024-04-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Chemical Heat Storage Mechanism in Alkaline Earth Metal Oxide: Ab Initio Modeling of the Initial Hydration Reaction on MgO(001) Surface\",\"authors\":\"Toyokazu Ishida*, and , Kazuya Ishimura, \",\"doi\":\"10.1021/acs.jpcc.3c08408\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Effective management of reusable waste heat is one of the most important technologies for reducing energy consumption and promoting energy conservation. In particular, chemical heat storage is a practical solution to the spatial and temporal mismatches of energy use. Among various chemical reactions, the reversible hydration process of periclase (MgO) to brucite [Mg(OH)<sub>2</sub>] is a practical candidate for the future applications in various social systems. In this article, in order to clarify the atomistic reaction mechanism of thermochemical heat storage in the MgO hydration reaction, we calculated potential energy profiles to understand the complex thermochemical reaction on the solid surface. By assuming a series of elementary reaction processes in the early stage of the surface reaction, we have practically estimated the reaction energetics for the hydration reaction in both the MgO(001) terrace and step geometries. First, we calculate two important thermodynamic parameters that characterize the thermochemical reaction (the reaction heat and the transition temperature). Our computational results show good agreement with the available thermodynamic data. Then, to clarify the nature of the thermochemical heat storage process, we introduce the 2D potential energy diagram, which is determined by the dislocation distance of the Mg atoms and the coordination (solvation) number of the Mg atoms. This 2D potential energy diagram clearly shows that the activation energy is highly dependent on the surface geometry of MgO, while an atomistic process in this rate-determining step (an extraction of Mg atoms from the inner layer to the surface aqueous layer) is exactly the same in both the terrace and step surface. These computational results lead to a simple guideline for the design of thermochemical heat storage systems.</p>\",\"PeriodicalId\":61,\"journal\":{\"name\":\"The Journal of Physical Chemistry C\",\"volume\":\"128 18\",\"pages\":\"7397–7407\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2024-04-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry C\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpcc.3c08408\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry C","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpcc.3c08408","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Chemical Heat Storage Mechanism in Alkaline Earth Metal Oxide: Ab Initio Modeling of the Initial Hydration Reaction on MgO(001) Surface
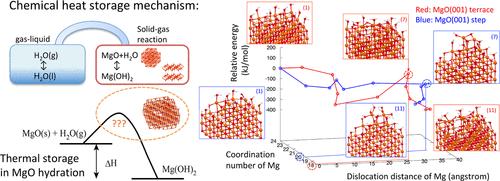
Effective management of reusable waste heat is one of the most important technologies for reducing energy consumption and promoting energy conservation. In particular, chemical heat storage is a practical solution to the spatial and temporal mismatches of energy use. Among various chemical reactions, the reversible hydration process of periclase (MgO) to brucite [Mg(OH)2] is a practical candidate for the future applications in various social systems. In this article, in order to clarify the atomistic reaction mechanism of thermochemical heat storage in the MgO hydration reaction, we calculated potential energy profiles to understand the complex thermochemical reaction on the solid surface. By assuming a series of elementary reaction processes in the early stage of the surface reaction, we have practically estimated the reaction energetics for the hydration reaction in both the MgO(001) terrace and step geometries. First, we calculate two important thermodynamic parameters that characterize the thermochemical reaction (the reaction heat and the transition temperature). Our computational results show good agreement with the available thermodynamic data. Then, to clarify the nature of the thermochemical heat storage process, we introduce the 2D potential energy diagram, which is determined by the dislocation distance of the Mg atoms and the coordination (solvation) number of the Mg atoms. This 2D potential energy diagram clearly shows that the activation energy is highly dependent on the surface geometry of MgO, while an atomistic process in this rate-determining step (an extraction of Mg atoms from the inner layer to the surface aqueous layer) is exactly the same in both the terrace and step surface. These computational results lead to a simple guideline for the design of thermochemical heat storage systems.
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


